私が誰でいつ何処に在るのか、過去も未来も、星座を配置する格子のように決定されていて、同時に別の星座の一つである。この痛みも直に形を変えていく。何に手を伸ばし、何に触れられるのか。知り得たことで書き換えらていくもの。視点が一つであろうと遍在しようと、同じだけの時間が必要になる。
過ちを繰り返しているのではない。正解を探しているのだ。

□ Diffusion analysis of single particle trajectories in a Bayesian nonparametrics framework
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/16/704049.full.pdf
This method is an infinite HMM (iHMM) within the general framework of Bayesian non-parametric models.
using a Bayesian nonparametric approach that allows the parameter space to be infinite-dimensional.
The Infinite Hidden Markov Model (iHMM) is a nonparametric model that has recently been applied to FRET data by Press ́e and coworkers to estimate the number of conformations of a molecule and simultaneously infer kinetic parameters for each conformational state.

□ Evaluation of simulation models to mimic the distortions introduced into squiggles by nanopore sequencers and segmentation algorithms
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0219495
Dynamic Time Warped-space averaging (DTWA) techniques can generate a consensus from multiple noisy signals without introducing key feature distortions that occur with standard averaging.
Z-normalized signal-to-noise ratios suggest intrinsic sensor limitations being responsible for half the gold standard and noisy squiggle Dynamic Time Warped-space differences.

□ Predicting Collapse of Complex Ecological Systems: Quantifying the Stability-Complexity Continuum
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/24/713578.full.pdf
Exploring the phase space as biodiversity and complexity are varied for interaction webs in which consumer-resource interactions are chosen randomly and driven by Generalized-Lotka-Volterra dynamics.
With this extended phase space and our construction of predictive measures based strictly on observable quantities, real systems can be better mapped – than using canonical measures by May or critical slowdown – for proximity to collapse and path through phase- space to collapse.
Allowing and accounting for these single- species extinctions reveals more detailed structure of the complexity-stability phase space and introduces an intermediate phase between stability and collapse – Extinction Continuum.
□ SHIMMER: Human Genome Assembly in 100 Minutes
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/17/705616.full.pdf
The most common approach to long-read assembly, using an overlap-layout-consensus (OLC) paradigm, requires all-to-all read comparisons, which quadratically scales in computational complexity with the number of reads.
Peregrine uses Sparse Hierarchical MiniMizERs (SHIMMER) to index reads thereby avoiding the need for an all-to-all read comparison step.
Peregrine maps the reads back to the draft contig and apply an updated FALCONsense algorithm to polish the draft contig.
This proposal for hyper-rapid assembly (i.e. in 100 minutes) overcomes quadratic scaling with a linear pre-processing step. the algorithmic runtime complexity to construct the SHIMMER index is O(GC) or O(NL).

□ MCtandem: an efficient tool for large-scale peptide identification on many integrated core (MIC) architecture
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2980-5
MCtandem, an efficient tool for large-scale peptide identification on Intel Many Integrated Core (MIC) architecture.
execute the MCtandem for a very large dataset on an MIC cluster (a component of the Tianhe-2 supercomputer) and achieved much higher scalability than in a benchmark MapReduce-based programs, MR-Tandem.

□ Possibility of group consensus arises from symmetries within a system
>> https://aip.scitation.org/doi/10.1063/1.5098335
an alternative type of group consensus is achieved for which nodes that are “symmetric” achieve a common final state.
The dynamic behavior may be distinct between nodes that are not symmetric.
a method derived using the automorphism group of the underlying graph which provides more granular information that splits the dynamics of consensus motion from different types of orthogonal, cluster breaking motion.


□Biophysics and population size constrains speciation in an evolutionary model of developmental system drift
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007177
The degree of redundancy can be represented as the “sequence entropy”, corresponding to the log of the number of genotypes corresponding to a given phenotype, in analogy to the similar expression in statistical mechanics.
explore a theoretical framework to understand how incompatibilities arise due to developmental system drift, using a tractable biophysically inspired genotype-phenotype for spatial gene expression.
The model allows for cryptic genetic variation and changes in molecular phenotypes while maintaining organismal phenotype under stabilising selection.
□ TWO-SIGMA: a novel TWO-component SInGle cell Model-based Association method for single-cell RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/22/709238.full.pdf
The first component models the drop-out probability with a mixed-effects logistic regression, and the second component models the (conditional) mean read count with a mixed-effects negative binomial regression.
Simulation studies and real data analysis show advantages in type-I error control, power enhancement, and parameter estimation over alternative approaches including MAST and a zero-inflated negative binomial model without random effects.

□ Mathematical modeling with single-cell sequencing data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/22/710640.full.pdf
building mathematical models of cell state-transitions with scRNA-seq data with hematopoeisis as a model system; by solving partial differential equations on a graph representing discrete cell state relationships, and by solving the equations on a continuous cell state-space.
calibrate model parameters from single or multiple time-point single-cell sequencing data, and examine the effects of data processing algorithms on the model calibration and predictions.
developing quantities, such as index of critical state transitions, in the phenotype space that could be used to predict forthcoming major alterations in development, and to be able to infer the potential landscape directly from the RNA velocity vector field.
□ At the edge of chaos: Recurrence network analysis of exoplanetary observables
>> https://phys.org/news/2019-07-edge-chaos-method-exoplanet-stability.html
an alternative method to perform the stability analysis of exoplanetary systems that requires only a scalar time series of the measurements, e.g., RV, transit timing variation (TTV), or astrometric positions.
The fundamental concept of Poincaré recurrences in closed Hamiltonian systems and the powerful techniques of nonlinear time series analysis combined with complex network representation allow us to investigate the underlying dynamics without having the equations of motion.
□ ATEN: And/Or Tree Ensemble for inferring accurate Boolean network topology and dynamics
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz563/5542393
a Boolean network inference algorithm which is able to infer accurate Boolean network topology and dynamics from short and noisy time series data.
ATEN algorithm can infer more accurate Boolean network topology and dynamics from short and noisy time series data than other algorithms.
□ BJASS: A new joint screening method for right-censored time-to-event data with ultra-high dimensional covariates
>> https://journals.sagepub.com/doi/10.1177/0962280219864710
a new sure joint screening procedure for right-censored time-to-event data based on a sparsity-restricted semiparametric accelerated failure time model.
BJASS consists of an initial screening step using a sparsity-restricted least-squares estimate based on a synthetic time variable and a refinement screening step using a sparsity-restricted least-squares estimate with the Buckley-James imputed event times.

□ Simulating astrophysical kinetics in space and in the laboratory
>> https://aip.scitation.org/doi/10.1063/1.5120277
Plasma jets are really important in astrophysics since they are associated with some of the most powerful and intriguing cosmic particle accelerators.
the particle spectra and acceleration efficiency predicted by these simulations can guide the interpretation of space and astronomical observations in future studies.


□ Efficient de novo assembly of eleven human genomes using PromethION sequencing and a novel nanopore toolkit
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/26/715722.full.pdf
To assemble these data they introduce new computational tools: Shasta - a de novo long read assembler, and MarginPolish & HELEN - a suite of nanopore assembly polishing algorithms.
On a single commercial compute node Shasta can produce a complete human genome assembly in under six hours, and MarginPolish & HELEN can polish the result in just over a day, achieving 99.9% identity (QV30) for haploid samples from nanopore reads alone.

□ On the discovery of population-specific state transitions from multi-sample multi-condition single-cell RNA sequencing data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/26/713412.full.pdf
Statistical power to detect changes in cell states also relates to the depth of sequencing per cell.
surveying the methods available to perform cross-condition differential state analyses, including cell-level mixed models and methods based on aggregated “pseudobulk” data.
□ Assessing key decisions for transcriptomic data integration in biochemical networks
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007185
compared 20 decision combinations using a transcriptomic dataset across 32 tissues and the definition of which reaction may be considered as active (reactions of the GEM with a non-zero expression level after overlaying the data) is mainly influenced by thresholding approach.
these decisions incl how to integrate gene expression levels using the Boolean relationships between genes, the selection of thresholds on expression data to consider the associated gene as “active” or “inactive”, and the order in which these steps are imposed.
□ Bayesian Correlation is a robust similarity measure for single cell RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/26/714824.full.pdf
Bayesian correlations are more reproducible than Pearson correlations. Compared to Pearson correlations, Bayesian correlations have a smaller dependence on the number of input cells.
And the Bayesian correlation algorithm assigns high similarity values to genes with a biological relevance in a specific population.

□ geneCo: A visualized comparative genomic method to analyze multiple genome structures
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz596/5539862
A visualization and comparative genomic tool, geneCo, is proposed to align and compare multiple genome structures resulting from user-defined data in the GenBank file format.
Information regarding inversion, gain, loss, duplication, and gene rearrangement among the multiple organisms being compared is provided by geneCo.
□ BioNorm: Deep learning based event normalization for the curation of reaction databases
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz571/5539693
BioNorm considers event normalization as a paraphrase identification problem. It represents an entry as a natural language statement by combining multiple types of information contained in it.
Then, it predicts the semantic similarity between the natural language statement and the statements mentioning events in scientific literature using a long short-term memory recurrent neural network (LSTM).
□ Magic-BLAST: an accurate RNA-seq aligner for long and short reads
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2996-x
Magic-BLAST is the best at intron discovery over a wide range of conditions and the best at mapping reads longer than 250 bases, from any platform.
As demonstrated by the iRefSeq set, only Magic-BLAST, HISAT2 with non-default parameters, STAR long and Minimap2 could align very long sequences, even if there were no mismatches.

□ GARDEN-NET and ChAseR: a suite of tools for the analysis of chromatin networks
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/28/717298.full.pdf
GARDEN-NET allows for the projection of user-submitted genomic features on pre-loaded chromatin interaction networks exploiting the functionalities of the ChAseR package to explore the features in combination with chromatin network topology.
ChAseR provides extremely efficient calculations of ChAs and other related measures, including cross-feature assortativity, local assortativity defined in linear or 3D space and tools to explore these patterns.

□ KDiffNet: Adding Extra Knowledge in Scalable Learning of Sparse Differential Gaussian Graphical Models
>> https://www.biorxiv.org/content/10.1101/716852v1
integrating different types of extra knowledge for estimating the sparse structure change between two p-dimensional Gaussian Graphical Models (i.e. differential GGMs).
KDiffNet incorporates Additional Knowledge in identifying Differential Networks via an Elementary Estimator.
a novel hybrid norm as a superposition of two structured norms guided by the extra edge information and the additional node group knowledge, and solved through a fast parallel proximal algorithm, enabling it to work in large-scale settings.
□ Multi-scale bursting in stochastic gene expression
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/28/717199.full.pdf
a stochastic multi-scale transcriptional bursting model, whereby a gene fluctuates between three states: two permissive states and a non-permissive state.
the time-dependent distribution of mRNA numbers is accurately approximated by a telegraph model with a Michaelis-Menten like dependence of the effective transcription rate on polymerase abundance.
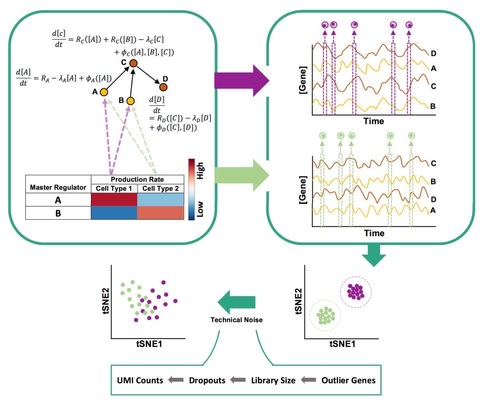
□ SERGIO: A single-cell expression simulator guided by gene regulatory networks
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/28/716811.full.pdf
SERGIO, a simulator of single-cell gene expression data that models the stochastic nature of transcription as well as linear and non-linear influences of multiple transcription factors on genes according to a user-provided gene regulatory network.
SERGIO is capable of simulating any number of cell types in steady-state or cells differentiating to multiple fates according to a provided trajectory, reporting both unspliced and spliced transcript counts in single-cells.

□ DeepHiC: A Generative Adversarial Network for Enhancing Hi-C Data Resolution
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/29/718148.full.pdf
Empowered by adversarial training, DeepHic can restore fine-grained details similar to those in high-resolution Hi-C matrices, boosting accuracy in chromatin loops identification and TADs detection.
DeepHiC- enhanced data achieve high correlation and structure similarity index (SSIM) compared with original high-resolution Hi-C matrices.
DeepHiC is a GAN model that comprises a generative network called generator and a discriminative network called discriminator.

□ OPERA-MS: Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes
>> https://www.nature.com/articles/s41587-019-0191-2
OPERA-MS integrates assembly-based metagenome clustering with repeat-aware, exact scaffolding to accurately assemble complex communities.
OPERA-MS assembles metagenomes with greater base pair accuracy than long-read (>5×; Canu), higher contiguity than short-read (~10× NGA50; MEGAHIT, IDBA-UD, metaSPAdes) and fewer assembly errors than non-metagenomic hybrid assemblers (2×; hybridSPAdes).
OPERA-MS provides strain-resolved assembly in the presence of multiple genomes of the same species, high-quality reference genomes for rare species with ~9× long-read coverage and near-complete genomes with higher coverage.
□ RITAN: rapid integration of term annotation and network resources
>> https://peerj.com/articles/6994/
RITAN is a simple knowledge management system that facilitates data annotation and hypothesis exploration—activities that are nor supported by other tools or are challenging to use programmatically.
RITAN allows annotation integration across many publically available resources; thus, it facilitates rapid development of novel hypotheses about the potential functions achieved by prioritized genes and multiple-testing correction across all resources used.
RITAN leverages multiple existing packages, extending their utility, including igraph and STRINGdb. Enrichment analysis currently uses the hypergeometric test.

□ Stochastic Lanczos estimation of genomic variance components for linear mixed-effects models
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2978-z
stochastic Lanczos derivative-free REML (SLDF_REML) and Lanczos first-order Monte Carlo REML (L_FOMC_REML), that exploit problem structure via the principle of Krylov subspace shift-invariance to speed computation beyond existing methods.
Both novel algorithms only require a single round of computation involving iterative matrix operations, after which their respective objectives can be repeatedly evaluated using vector operations.

□ IRESpy: an XGBoost model for prediction of internal ribosome entry sites
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2999-7
IRESpy, a machine learning model that combines sequence and structural features to predict both viral and cellular IRES, with better performance than previous models.
The XGBoost model performs better than previous classifiers, with higher accuracy and much shorter computational time.
□ ROBOT: A Tool for Automating Ontology Workflows
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3002-3
ROBOT (a recursive acronym for “ROBOT is an OBO Tool”) was developed to replace OWLTools and OORT with a more modular and maintainable code base.
ROBOT also helps guarantee that released ontologies are free of certain types of logical errors and conform to standard quality control checks, increasing the overall robustness and efficiency of the ontology development lifecycle.

□ Shiny-Seq: advanced guided transcriptome analysis
>> https://bmcresnotes.biomedcentral.com/articles/10.1186/s13104-019-4471-1
Shiny-Seq pipeline provides two different starting points for the analysis. First, the count table, which is the universal file format produced by most of the alignment and quantification tools.
Second, the transcript-level abundance estimates provided by ultrafast pseudoalignment tools like kallisto.
□ SIENA: Bayesian modelling to assess differential expression from single-cell data
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/30/719856.full.pdf
two novel approaches to perform DEG identification over single-cell data: extended Bayesian zero-inflated negative binomial factorization (ext-ZINBayes) and single-cell differential analysis (SIENA).
ext-ZINBayes adopts an existing model developed for dimensionality reduc- tion, ZINBayes. SIENA operates under a new latent variable model defined based on existing models.
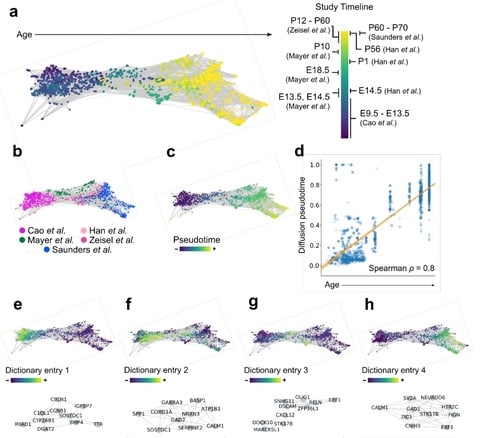
□ Coexpression uncovers a unified single-cell transcriptomic landscape
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/30/719088.full.pdf
a novel algorithmic framework that analyzes groups of cells in coexpression space across multiple resolutions, rather than individual cells in gene expression space, to enable multi-study analysis with enhanced biological interpretation.
This approach reveals the biological structure spanning multiple, large-scale studies even in the presence of batch effects while facilitating biological interpretation via network and latent factor analysis.
□ Framework for determining accuracy of RNA sequencing data for gene expression profiling of single samples
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/30/716829.full.pdf
This strategy for measuring RNA-Seq data content and identifying thresholds could be applied to a clinical test of a single sample, specifying minimum inputs and defining the sensitivity and specificity.
estimating a sample sequenced to the depth of 70 million total reads will typically have sufficient data for accurate gene expression analysis.

□ Graphmap2 - splice-aware RNA-seq mapper for long reads
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/30/720458.full.pdf
This extended version uses the same five-stage ‘read-funneling’ approach as the initial version and adds upgrades specific for mapping RNA reads.
With high number of reads mapped to the same reference region by Graphmap2 and Minimap2 for which no previous annotation exists, as well as high number of donor-acceptor splice sites in alignments of these reads,
Graphmap2 alignments provide indication that these alignments could belong to previously unknown genes.
□ DeLTA: Automated cell segmentation, tracking, and lineage reconstruction using deep learning
>> https://www.biorxiv.org/content/biorxiv/early/2019/07/31/720615.full.pdf
The framework is not constrained to a particular experimental set up and has the potential to generalize to time-lapse images of other organisms or different experimental configurations.
DeLTA (Deep Learning for Time-lapse Analysis), an image processing tool that uses two U-Net deep learning models consecutively to first segment cells in microscopy images, and then to perform tracking and lineage reconstruction.
□ Gaussian Mixture Copulas for High-Dimensional Clustering and Dependency-based Subtyping
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz599/5542387
HD-GMCM outperforms state-of-the-art model-based clustering methods, by virtue of modeling non-Gaussian data and being robust to outliers through the use of Gaussian mixture copulas.

□ PathwayMatcher: proteoform-centric network construction enables fine-granularity multiomics pathway mapping
>> https://academic.oup.com/gigascience/article/8/8/giz088/5541632
PathwayMatcher enables refining the network representation of pathways by including proteoforms defined as protein isoforms with posttranslational modifications.
PathwayMatcher is not developed as a mechanism inference or validation tool, but as a hypothesis generation tool.
□ ReadsClean: a new approach to error correction of sequencing reads based on alignments clustering
>> https://arxiv.org/pdf/1907.12718.pdf
The algorithm is implemented in ReadsClean program, which can be classified as multiple sequence alignment-based.
ReadsClean clustering approach is very useful for error correction in genomes containing multiple groups of repeated sequences, when the correction must be done within the corresponding repeat cluster.