










□ Haplotype-aware diplotyping from noisy long reads
>> https://www.readcube.com/articles/10.1186/s13059-019-1709-0
for contemporary long read technologies, read-based phase inference can be simultaneously combined with the genotyping process for SNVs to produce accurate diplotypes and to detect variants in regions not mappable by short reads.
using haplotype information during genotyping makes it possible to detect uncertainties and potentially compute more reliable genotype predictions.
formulated a novel statistical framework based upon hidden Markov models (HMMs) to analyze long-read sequencing data. a probabilistic model for diplotype inference, and primarily, to find maximum posterior probability genotypes.
□ BioDiscML: Large-Scale Automatic Feature Selection for Biomarker Discovery in High-Dimensional OMICs Data
>> https://www.frontiersin.org/articles/10.3389/fgene.2019.00452/full
BioDiscML exploits various feature selection procedures to produce signatures associated to machine learning models that will predict efficiently a specified outcome.
BioDiscML uses a large variety of machine learning algorithms to select the best combination of biomarkers for predicting categorical or continuous outcomes from highly unbalanced datasets.
BioDiscML also retrieves correlated biomarkers not included in the final model to better understand the signature. The software has been implemented to automate all machine learning steps, incl. data pre-processing, feature selection, model selection, and performance evaluation.
□ Generic Repeat Finder: a high-sensitivity tool for genome-wide de novo repeat detection
>> http://www.plantphysiol.org/content/early/2019/05/31/pp.19.00386
As a generic bioinformatics tool in repeat finding implemented as a parallelized C++ program, GRF was faster and more sensitive than existing inverted repeat/MITE detection tools based on numerical approaches (i.e., detectIR and detectMITE).
GRF sensitively identifies terminal inverted repeats (TIRs), terminal direct repeats (TDRs), and interspersed repeats that bear both inverted/direct repeats.
Generic Repeat Finder (GRF), a tool for genome-wide repeat detection based on fast, exhaustive numerical calculation algorithms integrated with optimized dynamic programming strategies.
□ DeepSymmetry: Using 3D convolutional networks for identification of tandem repeats and internal symmetries in protein structures
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz454/5510549
DeepSymmetry is designed to identify tandem repeat proteins, proteins with internal symmetries, symmetries in the raw density maps, their symmetry order, and also the corresponding symmetry axes.
Detection of symmetry axes is based on learning six-dimensional Veronese mappings of 3D vectors, and the median angular error of axis determination is less than one degree.
And demonstrate the capabilities of DeepSymmetry on benchmarks with tandem repeated proteins and also with symmetrical assemblies.
□ eFORGE v2.0: updated analysis of cell type-specific signal in epigenomic data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz456/5510552
significantly updated and improved version of eFORGE that can analyse both EPIC and 450k array data. New features include analysis of chromatin states, TF motifs and DNase I footprints, providing tools for EWAS interpretation and epigenome editing.
□ NX4: An alternative to genomic large genomic alignments: A visualization of large multiple sequence alignments
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz457/5510560
NX4, a Multiple Sequence Alignments visualization tool which can handle genome alignments comprising thousands of sequences.
NX4 calculates the frequency of each nucleotide along the alignment and visually summarizes the results using a color-blind friendly palette that helps identifying regions of high genetic diversity.
X4 also provides the user with additional assistance in finding these regions with a “focus + context” mechanism that uses a line chart of the Shannon entropy across the alignment.

□ FC-R2 atlas: Recounting the FANTOM Cage Associated Transcriptome
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/04/659490.full.pdf
FC-R2, a comprehensive expression atlas across a broadly-defined human transcriptome, inclusive of over 100,000 coding and non-coding genes as described by the FANTOM CAGE-Associated Transcriptome (FANTOM-CAT).
FC-R2 Atlas atlas greatly extends the gene annotation used in the original recount2 resource, and will empower other researchers to investigate the roles of both known genes and recently described lncRNAs.
□ Integration of genomic variation and phenotypic data using HmtPhenome
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/04/660282.full.pdf
HmtPhenome, a new web resource that aims at providing a visual network of connections among variants, genes, phenotypes and diseases having any level of involvement in the mitochondrial functionality.
Data are collected from several third party resources and aggregated on the fly, allowing users to clearly identify interesting relations between the involved entities.
Tabular data with additional hyperlinks are also included in the output returned by HmtPhenome, so that users can extend their analysis with further information from external resources.
□ A DNA-based synthetic apoptosome
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/04/660183.full.pdf
The spatial organization of proteins in these higher-order signaling complexes facilitates proximity-driven activation and inhibition events, allowing tight regulation of the flow of information.
the programmability and modularity of DNA origami as a controllable molecular platform for studying protein-protein interactions involved in intracellular signaling.
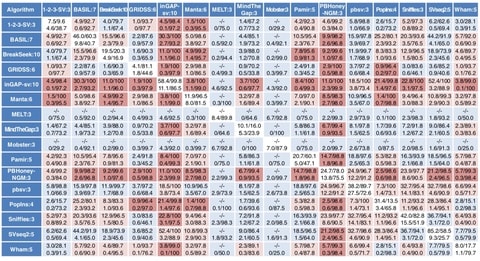
□ Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1720-5
systematic evaluation for overlapping calls from each combination of algorithm pairs demonstrates that several specific pairs of algorithms give a higher precision and recall for specific SV types and size ranges compared with other pairs.
enumerate potential good algorithms for each SV category, among which GRIDSS, Lumpy, SVseq2, SoftSV, Manta, and Wham are better algorithms in deletion or duplication categories.
□ BiosyntheticSPAdes: Reconstructing Biosynthetic Gene Clusters From Assembly Graphs
>> https://genome.cshlp.org/content/early/2019/06/03/gr.243477.118.full.pdf
While it is difficult to predict Biosynthetic Gene Clusters spanning multiple contigs, the structure of the genome assembly graph often provides clues on how to combine multiple contigs into segments encoding long BGCs.
biosyntheticSPAdes, a tool for predicting Biosynthetic Gene Clusters in assembly graphs and demonstrate that it greatly improves the reconstruction of BGCs from genomic and metagenomics datasets.

□ Space is the Place: Effects of Continuous Spatial Structure on Analysis of Population Genetic Data
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/03/659235.full.pdf
the combination of spatially autocorrelated environments and limited dispersal causes genome-wide association studies to identify spurious signals of genetic association with purely environmentally determined phenotypes,
and that this bias is only partially corrected by regressing out principal components of ancestry, and discuss the relevance of our simulation results for inference from genetic variation in real organisms.

□ A combination of transcription factors mediates inducible interchromosomal contacts
>> https://elifesciences.org/articles/42499
a method that enables the simultaneous testing of hundreds of cis or trans-acting mutations for their effects on a chromosomal contact of interest.
The MAP-C method will allow researchers to better understand which transcription factors control how DNA is folded inside the cell, and which mutations change this folding.

□ Composite Metagenome-Assembled Genomes Reduce the Quality of Public Genome Repositories
>> https://mbio.asm.org/content/10/3/e00725-19
A pangenomic analysis of the original and refined MAG III.A genomes with other publicly available Saccharibacteria genomes showed a 7-fold increase in the number of single-copy core genes.
These findings demonstrate the potential implications of composite MAGs in comparative genomics studies where single-copy core genes are commonly used to infer diversity, phylogeny, and taxonomy.
Composite MAGs can also lead to inaccurate ecological insights through inflated abundance and prevalence estimates.

□ Scaling tree-based automated machine learning to biomedical big data with a feature set selector
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz470/5511404
FSS increases TPOT’s efficiency in application on big data by slicing the entire dataset into smaller sets of features and allowing genetic programming to select the best subset in the final pipeline.
TPOT-FSS significantly outperforms a tuned XGBoost model and standard TPOT implementation.
□ Determining a random Schrödinger operator: both potential and source are random
>> https://arxiv.org/pdf/1906.01240v1.pdf
an inverse scattering problem associated with a Schrödinger system where both the potential and source terms are random and unknown.
The ergodicity is used to establish the single realization recovery. The asymptotic arguments in our study are based on the theories of pseudodifferential operators and microlocal analysis.
□ Properties of mean dimension and metric mean dimension coming from the topological entropy
>> https://arxiv.org/pdf/1905.13299v1.pdf
Single continuous map are classified by topological conjugacy. Non-autonomous dynamical systems are classified by uniform equiconjugacy.
the mean dimension and the metric mean dimension for non-autonomous dynamical systems and for single continuous maps, some properties hold for both non-autonomous and autonomous systems.

□ SANTA-SIM: simulating viral sequence evolution dynamics under selection and recombination
>> https://academic.oup.com/ve/article/5/1/vez003/5372481
Simulations of evolutionary histories in population genetics can be categorized either as forward-in-time or backwards-in-time (coalescent) genealogical models.
SANTA-SIM implements an individual-based, discrete-generation, and forwards-time simulator for molecular evolution of genetic data in a finite population.
□ Aquila: diploid personal genome assembly and comprehensive variant detection based on linked reads
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/05/660605.full.pdf
Aquila achieves contiguity for both haplotypes on a genome-wide scale, and its phasing nature guarantees a real haplotype- resolved assembly instead of a haploid consensus assembly.
Over 98% of a human Aquila- assembled genome is diploid, facilitating detection of the most prevalent types of human genetic variation, including SNPs, small indels, and structural variants (SVs), in all but the most difficult regions.
All heterozygous variants are phased in blocks that can approach arm-level length. The final output of Aquila is a diploid and phased personal genome sequence, and a phased VCF file that also contains homozygous and a few unphased heterozygous variants.

□ iruka_okeke’s talk: Remarkable progress made in AMR surveillance in Nigeria .. from zero (no national action plan) to hero (training, capacity building, quality assurance, genomic surveillance) #ABPHM19 via @EsTeeTorok
□ IMPRes: A Dynamic Programming Approach to Integrate Gene Expression Data and Network Information for Pathway Model Generation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz467/5511838
IMPRes algorithm, a new step-wise active pathway detection method using a dynamic programming approach.
starting from one or multiple seed genes, a shortest path algorithm is applied to detect downstream pathways that best explain the gene expression data.
□ Factor analysis for survival time prediction with informative censoring and diverse covariates
>> https://onlinelibrary.wiley.com/doi/full/10.1002/sim.8151
an integrative latent variable model that combines factor analysis for various data types and an exponential proportional hazards (EPH) model for continuous survival time with informative censoring.
Integrative modeling is sensible, as the underlying hypothesis is that joint analysis of multiple covariates provides greater explanatory power than separate analyses.
□ methyl-ATAC-seq measures DNA methylation at accessible chromatin
>> https://genome.cshlp.org/content/early/2019/06/03/gr.245399.118
methyl-ATAC-seq (mATAC-seq), which implements modifications to ATAC-seq, including subjecting the output to BS-seq.
Merging these assays into a single protocol identifies the locations of open chromatin and reveals, unambiguously, DNA methylation state of the underlying DNA. Such combinatorial methods eliminate the need to perform assays independently and infer where features are coincident.
□ Single-cell transcriptomics unveils gene regulatory network plasticity
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1713-4
a conceptually different computational framework based on a holistic view, where single-cell datasets are used to infer global, large-scale regulatory networks.
correlation metrics that are specifically tailored to single-cell data, and then generate, validate, and interpret single-cell-derived regulatory networks from organs and perturbed systems.

□ BAGSE: a Bayesian hierarchical model approach for gene set enrichment analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/06/662171.full.pdf
BAGSE is built on a natural Bayesian hierarchical model and fully accounts for the uncertainty embedded in the association evidence of individual genes.
BAGSE performs both enrichment hypothesis testing and quantification. It requires gene-level association evidence (in forms of either z-scores or estimated effect sizes with corresponding standard errors) and pre-defined gene set annotations as input.
BAGSE can simultaneously handle multiple and/or mutually non-exclusive gene set definitions, a feature currently missing from the existing methods.
□ LIGER: Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity
>> https://www.cell.com/cell/fulltext/S0092-8674(19)30504-5
□ LIGER (Linked Inference of Genomic Experimental Relationships): integrating and analyzing multiple single-cell datasets
>> https://macoskolab.github.io/liger/
LIGER, an algorithm that delineates shared and dataset-specific features of cell identity.
LIGER relies on integrative non-negative matrix factorization to identify shared and dataset-specific factors.
□ Seurat v3: Comprehensive Integration of Single-Cell Data
>> https://www.cell.com/cell/fulltext/S0092-8674(19)30559-8
a strategy to “anchor” diverse datasets together, enabling us to integrate single-cell measurements not only across scRNA-seq technologies, but also across different modalities.
Seurat v3 provides a strategy for the assembly of harmonized references and transfer of information across datasets.
□ hypeR: An R Package for Geneset Enrichment Workflows
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/06/656637.full.pdf
hypeR is a comprehensive R package for geneset enrichment workflows that offers multiple enrichment, visualization, and sharing methods in addition to novel features such as hierarchical geneset analysis and built-in markdown reporting.
A hyp object contains all information relevant to the enrichment analysis, including a data frame of results, enrich- ment plots for each geneset tested, as well as the arguments used to perform the analysis.
□ Alvis: a tool for contig and read ALignment VISualisation and chimera detection
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/06/663401.full.pdf
Alvis, a simple command line tool that can generate visualisations for a number of common alignment analysis tasks.
Alvis is a fast and portable tool that accepts input in the most common alignment formats and will output production ready vector images.
Alvis will highlight potentially chimeric reads or contigs, a common source of misassemblies.
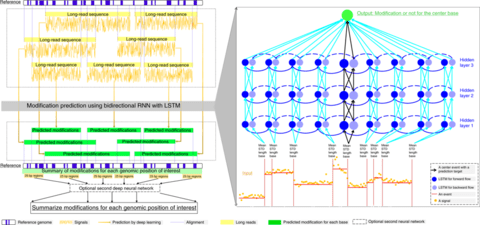
□ DeepMod: Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data
>> https://www.nature.com/articles/s41467-019-10168-2
DeepMod, a bidirectional recurrent neural network (RNN) with long short-term memory (LSTM) to detect DNA modifications.
DeepMod is a well-trained bidirectional recurrent neural network (RNN) with LSTM units, which takes signal mean, standard deviation, and the number of signals of an event together with base information in the reference genome of an event and its neighbors as input.
Then, after anchoring events with a reference genome based on the alignment of long reads, predicted modification summary for reference positions of interest can be generated in a BED format.
The prediction of DNA modification by DeepMod is thus strand-sensitive and has single-base resolution.

□ Paired-end Mappability of Transposable Elements in the Human Genome
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/06/663435.full.pdf
a few of all TE loci in the genome have been observed to be transcriptionally active depending on the tissue and developmental time point, and the majority of the uniquely mappable TE loci we have identified may be biologically irrelevant.
This paired-end mappability analysis suggests that longer paired-end read libraries can be confidently mapped to repetitive regions and specifically to the locus-level of the majority of TEs.

□ Atos to deliver most powerful supercomputer in Norway to national e-infrastructure provider Uninett Sigma2” BullSequana XH2000, 172032 cores, AMD EPYC processors, 5.9 Pflops.
□ RAISS: Robust and Accurate imputation from Summary Statistics
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz466/5512360
RAISS is a python package enabling the imputation of SNP summary statistics from the neighboring SNPs by taking advantage of the Linkage desiquilibrium.
While methods for the imputation of summary statistics exist, they lack precision for genetic variants with small effect size. This is benign for univariate analyses where only variants with large effect size are selected a posteriori.
RAISS is a new approach that improve the existing imputation methods and reach a precision suitable for multi-trait analyses, the resulting methodology specially designed to efficiently impute multiple GWAS in parallel.
□ DepthFinder: A Tool to Determine the Optimal Read Depth for Reduced-Representation Sequencing
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz473/5512354
Restriction site associated sequencing (RSAS) methodologies have been widely used for rapid and cost-effective discovery of SNPs and for high-throughput genotyping in a wide range of species.
DepthFinder is designed to estimate the required read counts for RSAS methods that cover a range of different biological (genome size, level of genome complexity, level of DNA methylation and ploidy) and technical (library preparation protocol and sequencing platform) factors.

□ Sparse discriminative latent characteristics for predicting cancer drug sensitivity from genomic features
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006743
a sparse multitask regression model which learns discriminative latent characteristics that predict drug sensitivity and are associated with specific molecular features.
Using Bayesian nonparametrics to automatically infer the appropriate number of these latent characteristics.
This approach is closely related to Kernelized Bayesian Multitask Learning. Sparse Cauchy priors are used to select features and a Dirichlet prior is used over a parameter vector that selects predictive views.
□ A versatile method for circulating cell-free DNA methylome profiling by reduced representation bisulfite sequencing
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/07/663195.full.pdf
a novel sample preparation method for reduced representation bisulfite sequencing (RRBS), rigorously designed and customized for minute amounts of highly fragmented DNA.
cf-RRBS stands out as striking a particularly good balance between genome methylation coverage, reproducibility, ease of execution and affordability.

□ On the computability properties of topological entropy: a general approach
>> https://arxiv.org/pdf/1906.01745v1.pdf
The dynamics of symbolic systems, such as multidimensional subshifts of finite type or cellular automata, are known to be closely related to computability theory.
In analogy to effective subshifts, consider computable maps over effective compact sets in general metric spaces, and study the computability properties of their topological entropies.
□ Properties of the full random effect modelling approach with missing covariates
>> https://www.biorxiv.org/content/biorxiv/early/2019/06/07/656470.full.pdf
the full random effect model (FREM), the covariates are treated as observed data points and are modelled as random effects instead of being treated as error free explanatory variables whose impact of the model is estimated through fixed effect parameters.
while the bias in the parameter estimates increased in a similar fashion for the reference method, the full random effects approach provided unbiased estimates for all degrees of covariate missingness.
□ TPMCalculator: one-step software to quantify mRNA abundance of genomic features
>> https://academic.oup.com/bioinformatics/article/35/11/1960/5150437
TPMCalculator quantifies mRNA abundance directly from the alignments by parsing BAM files.
The input parameters are the same GTF files used to generate the alignments, and one or multiple input BAM file(s) containing either single-end or paired-end sequencing reads.
□ Lazer / LaSAGNA: High-Performance Computing Frameworks for Large-Scale Genome Assembly:
>> https://digitalcommons.lsu.edu/gradschool_dissertations/4942/
Lazer achieves both scalability and memory efficiency by using partitioned de Bruijn graphs.
By enhancing the memory-to-disk swapping and reducing the network communication in the cluster, Lazer can assemble large sequences such as human genomes (~400 GB) on just two nodes in 14.5 hours, and also scale up to 128 nodes in 23 minutes.
the first distributed 3rd generation sequence (3GS) assembler which uses a map-reduce computing paradigm and a distributed hash-map, both built on a high-performance networking middleware.
Using this assembler, we assembled an Oxford Nanopore human genome dataset (~150 GB) in just over half an hour using 128 nodes whereas existing 3GS assemblers could not assemble it because of memory and/or time limitations.
LaSAGNA is a new distributed GPU-accelerated NGS assembler, which can assemble large-scale sequence datasets using a single GPU by building string graphs from approximate all-pair overlaps in quasi-linear time.

□ APARENT: A Deep Neural Network for Predicting and Engineering Alternative Polyadenylation
>> https://www.cell.com/cell/fulltext/S0092-8674(19)30498-2
Trained a neural network to predict APA using data from over 3 million reporters, and Predicted and experimentally characterized over 12,000 human APA variants.
Visualizing features learned across all network layers reveals that APARENT recognizes sequence motifs known to recruit APA regulators, discovers previously unknown sequence determinants of 3′ end processing, and integrates these features into a comprehensive, cis-regulatory code.