









"If you want to go fast, go alone. If you want to go far, go together."
"Genomic data are inherently and insidiously NOT IID." (Independent and identically distributed random variables.)
- Katherine S. Pollard (Genome Informatics 2018)

□ DeepSequence: Deep generative models of genetic variation capture the effects of mutations:
>> https://www.nature.com/articles/s41592-018-0138-4
DeepSequence, a probabilistic model for sequence families, predicted the effects of mutations across a variety of deep mutational scanning experiments substantially better than existing methods based on the same evolutionary data. The model, learned in an unsupervised manner solely on the basis of sequence information, is grounded with biologically motivated priors, reveals the latent organization of sequence families, and can be used to explore new parts of sequence space.
□ DeepVariant: A universal SNP and small-indel variant caller using deep neural networks:
>> https://www.nature.com/articles/nbt.4235
DeepVariant is a deep convolutional neural network can call genetic variation in aligned next-generation sequencing read data by learning statistical relationships between images of read pileups around putative variant and true genotype calls. DeepVariant can learn to call variants in a variety of sequencing technologies and experimental designs, including deep whole genomes from 10X Genomics and Ion Ampliseq exomes. The learned model generalizes across genome builds and mammalian species, allowing nonhuman sequencing projects to benefit from the wealth of human ground-truth data.

□ BiT-STARR-seq: High throughput characterization of genetic effects on DNA:protein binding and gene transcription:
>> http://genome.cshlp.org/content/early/2018/09/25/gr.237354.118.full.pdf
BiT-STARR-seq (Biallelic Targeted STARR-seq), a streamlined protocol for a high-throughput reporter assay, that identifies allele-specific expression (ASE) while accounting for PCR duplicates through unique molecular identifiers. Unlike STARR-seq, this method does not require preparation of DNA regions for use in the assay, such as whole genome fragmentation, or targeting regions, while, similar to STARR-seq, it requires only a single cloning and transformation step.

□ Trans effects on gene expression can drive omnigenic inheritance:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/24/425108.full.pdf
a formal model in which genetic contributions to complex traits can be partitioned into direct effects from core genes, and indirect effects from peripheral genes acting as trans-regulators. if the core genes for a trait are co-regulated – as seems likely – then the effects of peripheral variation can be amplified by these co-regulated networks such that nearly all of the genetic variance is driven by peripheral genes.

□ Identify tissue structure with Automatic Expression Histology using STARmap data:
>> http://www.nxn.se/valent/2018/9/25/identify-tissue-structure-with-automatic-expression-histology
Implemented in the SpatialDE package there is a Bayesian inference algorithm for learning posterior probabilities of the assignments z between genes and hidden spatial patterns that, together, give rise to spatial co-expression. STARmap (spatially-resolved transcript amplicon readout mapping) begins with labeling of cellular RNAs by pairs of DNA probes followed by enzymatic amplification so as to produce a DNA nanoball (amplicon), which eliminates background caused by mislabeling of single probes.

□ Mapping DNA replication with nanopore sequencing:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/26/426858.full.pdf
harnessing nanopore sequencing to study DNA replication genome-wide at the single-molecule level. Using in vitro prepared DNA substrates, characterize the effect of bromodeoxyuridine (BrdU) substitution for thymidine on the MinION nanopore electrical signal. implementing RepNano, a recurrent neural network with an architecture similar to DeepNano, to convert the raw current from nanopore experiments into a DNA sequence.
□ CaSpER: Identification, visualization and integrative analysis of CNV events in multiscale resolution using single-cell or bulk RNA sequencing data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/26/426122.full.pdf
CaSpER models the allele-based frequencies as a mixture of Gaussian distributions for identification and classification of genotype clusters. The shift in allelic signal is used to quantify the loss-of-heterozygosity (LOH) which is valuable for CNV identification. CaSpER uses Hidden Markov Models (HMM) to assign copy number states to regions. The multiscale nature of CaSpER enables comprehensive analysis of focal and large-scale CNVs and LOH segments.
□ Atlas-CNV: a validated approach to call Single-Exon CNVs in the eMERGESeq gene panel:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/27/422337.full.pdf
Atlas-CNV is validated as a method to identify exonic CNVs in targeted sequencing data generated in the clinical laboratory. The ExonQC and C-score assignment can reduce FDR (identification of targets with high variance) and improve calling accuracy of single-exon CNVs respectively. they proposed guidelines and criteria to identify high confidence single-exon CNVs.
□ Detection and Mitigation of Spurious Antisense RNA-seq Reads with RoSA:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/26/425900.full.pdf
RoSA (Removal of Spurious Antisense), detects the presence of high levels of spurious antisense read alignments in strand-specific RNA-Seq datasets. RoSA uses incorrectly spliced reads on the antisense strand and/or ERCC spike-ins (if present in the data) to calculate both global and gene-specific antisense correction factors.
□ Artificial Dilution Series: Novel Comparison of Evaluation Metrics for Gene Ontology Classifiers Reveals Drastic Performance Differences:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/27/427096.full.pdf
Artificial Dilution Series is the first method for testing various classifier evaluation metrics using real datasets as a platform for controlled amounts of embedded signal, and allows a simple testing of evaluation metrics to see how easily can separate different signal levels. This work proposes improved versions for some well-known evaluation metrics. The presented methods are also applicable to other areas of science where evaluation of prediction results is non-trivial.
□ plyranges: A grammar of genomic data transformation:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/26/327841.full.pdf
Genome Query Language and its distributed implementation GenAp which use a SQL-like syntax for fast retrieval of information of unprocessed sequencing data. Similarly, the Genometric Query Language (GMQL) implements a DSL for combining genomic datasets. a genomic DSL called plyranges that reformulates notions from existing genomic algebras and embeds them in R as a genomic extension of dplyr. By analogy, plyranges is to the genomic algebra, as dplyr is to the relational algebra.
□ GRID-seq assisted computational prediction of transcription factor binding motifs using multivariate mahalanobis distance analysis reveals that RNA-chromosomal interaction may act as a proxy indicator of true positive transcriptional activity.:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/27/429332.full.pdf
the global RNA interactions with DNA by deep sequencing (GRID-seq) approach, which allows a user to quantify genome wide binding of RNA to chromatin in a manner that results in data similar to what might be obtained from model-based analysis of ChIP-Seq (MACS). The aim of combining DeepBind and GRID-seq is to derive results that may assist researchers in study target selection. The desired outcome is achieved by simultaneously considering the influence that scores from GRID-seq and DeepBind has as a single distance function based on the distribution, and then ranking sequences that contain predicted binding motifs according to this new combined score.
□ SMARTer single cell total RNA sequencing:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/29/430090.full.pdf
The total RNA-seq method detects an equal or higher number of genes compared to classic polyA[+] RNA-seq, including novel and non-polyadenylated genes. The obtained RNA expression patterns also recapitulate the expected biological signal. Inherent to total RNA-seq, this method is also able to detect circular RNAs.

□ GEViT: A systematic method for surveying data visualizations and a resulting genomic epidemiology visualization typology:
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/bty832/5107018
GEViT consists of an initial literature analysis phase followed by a visualization analysis phase, resulting in a visualization design space in which images are classified according to their why and their how. The literature analysis phase automatically analyzes text from a corpus of research articles to identify the topic of a data visualization – why it was created – as assuming that different topics are likely to yield different visualization designs.
□ Enspara: Modeling molecular ensembles with scalable data structures and parallel computing:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/29/431072.full.pdf
enspara is a library for building Markov State Models at scale. a numpy-compatible implementation of the ragged array, which dramatically improved the memory footprint of Markov State Models-associated data. enspara also has turn-key sparse matrix usage. for users who wish to prototype entirely new Markov State Models estimation methods, any function or callable object is accepted as a builder, as long as it accepts a transition counts matrix C as input & returns a 2-tuple of transition probabilities & equilibrium probabilities.
information from a Markov State Model cannot be trivially substituted for frame-by-frame calculations. they also implement a function using cython and OpenMP that takes a trajectory of n features and returns a 4-dimensional joint counts array with dimension n × n × sn × sn. The value of returning this four-dimensional joint counts matrix is that it renders the problem embarrassingly parallel in the number of trajectories.
In enspara, a reference implementation of Correlation of All Rotameric and Dynamical States framework (CARDS). In brief, this method takes a series of molecular dynamics trajectories and computes the mutual information (MI) between all pairs of dihedral angle rotameric states, and between all pairs of dihedral angle order/disorder states. A dihedral angle is considered disordered if it frequently hops between rotameric states. This implementation parallelizes across cores on a single machine using the thread-parallelization.
□ MetaMap, an interactive webtool for the exploration of metatranscriptomic reads in human disease-related RNA-seq data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/30/425439.full.pdf
MetaMap applied the pipeline to close to 150 terabytes of publicly available raw RNA-seq data from >17,000 samples from >400 studies relevant to human disease using state-of-the-art high performance computing systems from the Leibniz Supercomputing Centre.
□ Vertical and horizontal integration of multi-omics data with miodin:
>> https://www.biorxiv.org/content/biorxiv/early/2018/09/30/431429.full.pdf
if(is(assayObj, "GenomicRatioSet")){
assayMetaData <- get(dataset, "assayMetaData", name = testAssay)
if("omega" %in% names(assayMetaData)){
omega = assayMetaData$omega[sampleTable$SampleName, ]
designMatrixCor <- cbind(designMatrix, omega)
□ The Chromium Single Cell ATAC (Assay for Transposase Accessible Chromatin) Solution
>> https://www.10xgenomics.com/solutions/single-cell-atac/#ATACseq
□ Challenges and guidelines toward 4D nucleome data and model standards
>> https://www.nature.com/articles/s41588-018-0236-3
it is now possible to build 3D models of how the genome folds within the nucleus and changes over time (4D). Because genome folding influences its function, this opens exciting new possibilities to broaden our understanding of the mechanisms that determine cell fate.
□ Everlasting Iatric Reader (Eir): Automatic Human-like Mining and Constructing Reliable Genetic Association Database with Deep Reinforcement Learning:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/05/434803.full.pdf
improve the reliability of text-mining by building a system that can directly simulate the behavior of a researcher, and develop corresponding methods, such as Bi-directional LSTM for text mining and Deep Q-Network for organizing behaviors. The immediate next-step plan is to train Eir for SNP-phenotype association with GWAS Catalog, and integrate these databases into GenAMap, a visual machine learning tool for GWASe, for validation purpose of GWAS results.
□ ewanbirney:
H3Africa refunded to $170 Million; 35 african countries, over 50 languages, many different legal systems, >100K people recruited, data deposition (collaboration with @EGAarchive) 380 trainees, 193 PhDs - Wow! #GA4GH2018
□ TransQST in a decade of public-private innovative medicines initiative (IMI) worth 3 billion €. Find out what 21 institutions from 9 countries do for developing safer drugs:
>> http://sbi.imim.es/data/imi-transqst.pdf
□ Nebula Genomics: Genomics startup bets on blockchain for data sharing platform:
>> https://www.mobihealthnews.com/content/genomics-startup-bets-blockchain-data-sharing-platform

□ AIDE: annotation-assisted isoform discovery and abundance estimation from RNA-seq data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/07/437350.full.pdf
Solving the isoform discovery problem in a stepwise and conservative manner, AIDE prioritizes the annotated isoforms and precisely identifies novel isoforms whose addition significantly improves the explanation of observed RNA-seq reads. AIDE learns gene and exon boundaries from annotations and also selectively borrows information from the annotated isoform structures using a stepwise likelihood-based selection approach.

□ scDesign: A statistical simulator for rational scRNA-seq experimental design:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/07/437095.full.pdf
scDesign is protocol- and data-adaptive. It learns scRNA-seq data characteristics from rapidly accumulating public scRNA-seq data generated under diverse settings. scDesign generates synthetic data that well mimic real scRNA-seq data under the same experimental settings, providing a basis for using its synthetic data to guide practical scRNA-seq experimental design.

□ Building gene regulatory networks from scATAC-seq and scRNA-seq using Linked Self-Organizing Maps:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/09/438937.full.pdf
Self-organizing maps (SOMs) are a type of artificial neural networks, also referred to as a Kohonen network. Combining the metaclusters from multiple SOMs as a pair-wise set generates a data-space that combines the properties from both without any assumptions about how the data relates to each-other. The ability of linked SOMs to detect emergent properties from multiple types of highly-dimensional genomic data with very different signal properties opens new avenues for integrative analysis of single-cells.
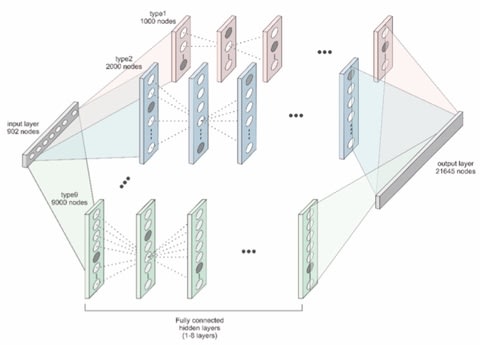
□ D-GPM: a deep learning method for gene promoter methylation inference:
>> https://www.biorxiv.org/content/biorxiv/early/2018/10/09/438218.full.pdf
D-GPM is a fully connected multi-layer perceptron with one output layer. All the hidden layers have the same number of hidden units. D-GPM contains 902 units in the input layer corresponding to the 902 landmark genes, and we also configure D-GPM with 21,645 units in the output layer analogous to the 21,645 target genes.










※コメント投稿者のブログIDはブログ作成者のみに通知されます