









(Created with Midjourney v7)
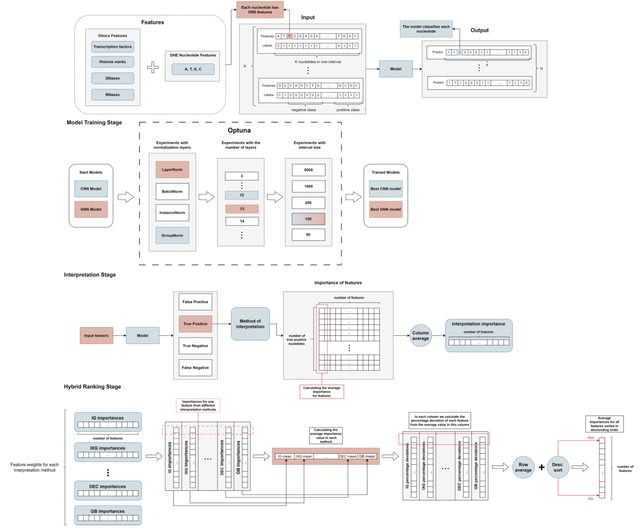
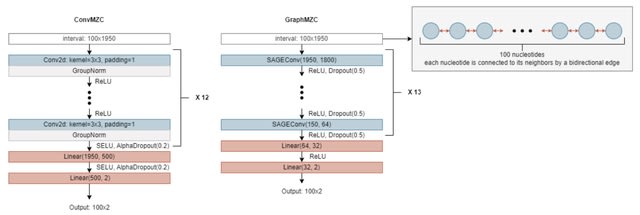

□ OmiXAI: An Ensemble XAI Pipeline for Interpretable Deep Learning in Omics Data
>> https://www.biorxiv.org/content/10.1101/2025.04.28.651097v1
OmiXAI, a pipeline integrating ensemble model-aware XAI methods. OmiXAI incorporates gradient-based techniques-incl. Integrated Gradients, InputXGradients, Guided Backpropagation, and Deconvolution (for CNNs and GNNs) — as well as Saliency Maps and GNNExplainer.
DNA sequences are one-hot encoded, omics features are integrated as additional features, and all data compressed using the SparseVector. All Target labels are binary encoded, with a value of 1 indicating the presence of a corresponding functional element within the DNA regions.
OmiXAI can compute gradients of the output with respect to the input tokens/features, and one can also compute gradients with respect to attention weights, which gives insights into which parts of the sequence the model is "looking at" to make predictions.


□ SCRIPT: predicting single-cell long-range cis-regulation based on pretrained graph attention networks
>> https://www.biorxiv.org/content/10.1101/2025.04.27.650894v1
SCRIPT (Single-cell Cis-regulatory Relationship Identifier based on Pre-Trained graph attention networks) leverages graph causal attention networks (GCATs) to simulate cis-transcriptional regulation, using chromatin accessibility of CREs measured by scATAC-seq to predict GE LVs.
Causal attention masks of GCATs, designed by incorporating empirical evidence from large-scale bulk Hi-C and eQTL datasets, enabling the modeling of cis-transcriptional regulation grounded in biological principles.
Second, SCRIPT employs a self-supervised graph autoencoder (SSGAE) pretrained on atlas-scale scATAC-seq data to comprehend the complex interactions between CREs across diverse tissues. It enables the model to generate effective CRE representations.
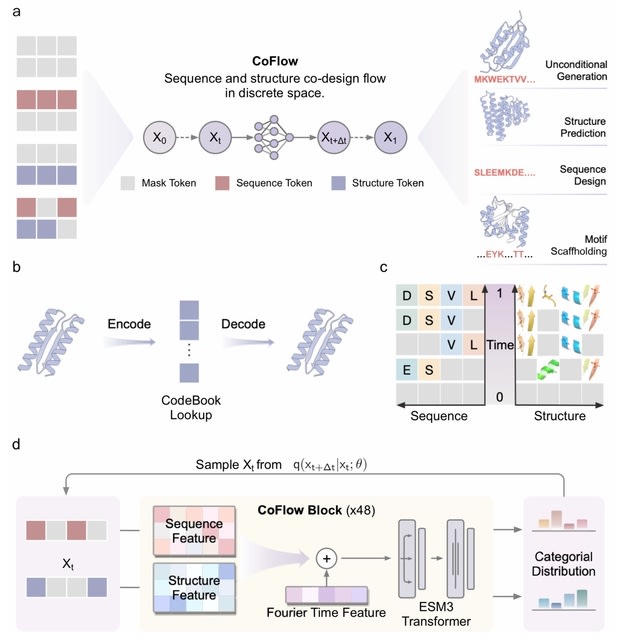
□ CoFlow: Co-Design protein sequence and structure in discrete space via generative flow
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf248/8123382
CoFlow leverages a joint generative flow to co-design protein sequences and backbone in discrete space. CoFlow operates within a probabilistic generative framework to model discrete distribution and sample protein instances.
CoFlow utilizes continuous-time Markov chains, providing greater flexibility than diffusion models and enabling more diverse and controllable sampling. The generative flow implements linear interpolation from noise to discrete tokens.
CoFlow incorporates the structure VQ-VAE from ESM3. CoFlow employs a bidirectional transformer enhanced with layer-wise Fourier time features to model sequence and structure within a unified latent space. The final outputs are two predicted categorical distributions.

□ scRegulate: Single-Cell Regulatory-Embedded Variational Inference of Transcription Factor Activity from Gene Expression
>> https://www.biorxiv.org/content/10.1101/2025.04.17.649372v1
scRegulate, a generative deep learning framework that leverages variational inference to infer TF activities while incorporating gene regulatory network (GRN) priors. scRegulate integrates structured biological constraints with a probabilistic latent space model.
scRegulate follows a three-phase approach: In the prior initialization phase, the weights connecting the TF activity layer to the output layer are initialized using the initial GRN, thereby enforcing known TF-gene regulatory interactions;
In the dynamic inference phase, regulatory weights are optimized, with prior constraints gradually relaxed to allow for the discovery of new TF-target interactions; In the cell-type-specific fine-tuning phase, GRNs are optimized per cell-type to refine TF activity patterns.
□ HDMA: Dissecting regulatory syntax in human development with scalable multiomics and deep learning
>> https://www.biorxiv.org/content/10.1101/2025.04.30.651381v1
the Human Development Multiomic Atlas (HDMA), a true multiomic, multi-organ single-cell atlas profiling chromatin accessibility and gene expression in 817,740 primary human fetal cells across 12 organs.
HDMA provides a foundational resource for decoding cis-regulatory syntax, linking sequence variation to gene regulation, and understanding how chromatin accessibility patterns drive human cell type diversity.
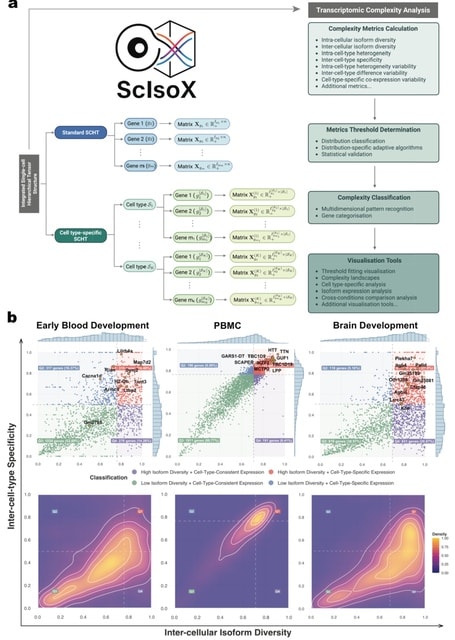
□ ScIsoX: A Multidimensional Framework for Measuring Transcriptomic Complexity in Single-Cell Long-Read Sequencing Data
>> https://www.biorxiv.org/content/10.1101/2025.04.28.650897v1
ScIsoX, a computational framework that implements (i) a novel Single-Cell Hierarchical Tensor (SCHT) data structure, ii) a comprehensive suite of analytical metrics, and (iii) visualisation tools for measuring transcriptomic complexity across multiple biological scales.
SCHT organises isoform-level count data into gene-specific sub-tensors, where each gene is represented by an individual count matrix containing isoform-by-cell expression values.
This partition-based design preserves the intrinsic hierarchy without resorting to extensive zero-padding, yielding a representation that is both biologically meaningful and computationally efficient.
SCHT is extended to include cell types as an additional dimension. Each count matrix contains only the cells belonging to that particular cell type expressing the gene, creating a multi-level hierarchy that elegantly captures gene-isoform-cell relationships.

□ EvoWeaver: large-scale prediction of gene functional associations from coevolutionary signals
>> https://www.nature.com/articles/s41467-025-59175-6
EvoWeaver provides as output its 12 predictions for signals of coevolution, and can optionally provide an ensemble prediction using built-in pretrained models. Functional associations often result in correlated gain/loss patterns on a reference phylogenetic tree.
EvoWeaver assesses the presence/absence patterns, correlation b/n gain/loss events, and distance b/n gain/loss events as signals of coevolution. It computes topological distance AWA correlation in patristic distances following dimensionality reduction using random projection.
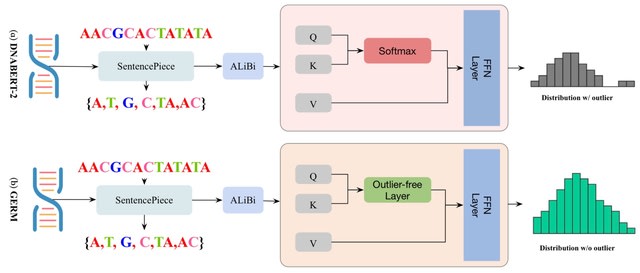
□ GERM: Fast and Low-Cost Genomic Foundation Models via Outlier Removal
>> https://arxiv.org/abs/2505.00598
GERM employs small-step continual learning within the outlier-free framework, leveraging original checkpoints to avoid retraining from scratch. Building on DNABERT-2, GERM incorporates QLoRA and LoFTQ, while integrating outlier suppression, OmniQuant for robust quantization.
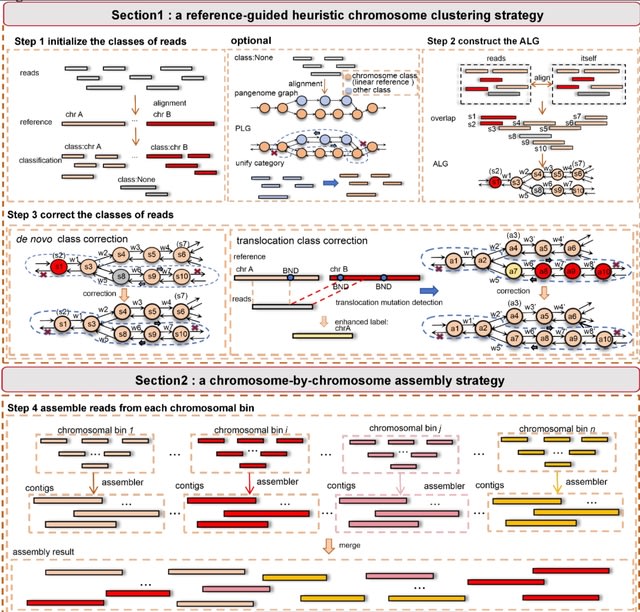
□ HiFiCCL: Reference-guided genome assembly at scale using ultra-low-coverage high-fidelity long-reads
>> https://www.biorxiv.org/content/10.1101/2025.04.20.649739v1
HiFiCCL designs a novel exclusively referenced-guided chromosome-by-chromosome assembly strategy, where whole-genome reads are partitioned by chromosome using the high-quality reference genome and then assembled individually.

□ RECUR: Identifying recurrent amino acid substitutions from multiple sequence alignments
>> https://www.biorxiv.org/content/10.1101/2025.04.29.651261v1
RECUR, a phylogenetic tool designed to address the gap by identifying recurrent substitutions, specifically parallel substitutions, that have occurred in a protein or codon multiple sequence alignment.
RECUR takes a multiple sequence alignment as input and identifies all recurrent sequence substitutions present within the evolutionary history of that alignment and their associated statistics.
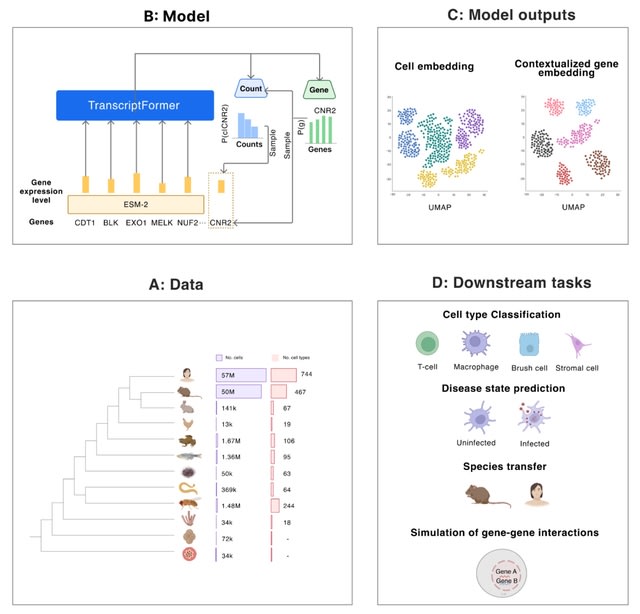
□ TranscriptFormer: A Cross-Species Generative Cell Atlas Across 1.5 Billion Years of Evolution: The TranscriptFormer Single-cell Model
>> https://www.biorxiv.org/content/10.1101/2025.04.25.650731v1
TranscriptFormer, a family of generative large-scale single-cell foundation models representing a digital cell atlas, trained on up to 112 million cells spanning 1.53 billion years of evolutionary history across species.
TranscriptFormer is a generative autoregressive joint model over genes and expression levels, with a transformer-based architecture incl. a coupling b/n gene and transcript heads, expression-aware multi-head self-attention, causal masking to capture transcript-level variability.

□ SGCRNA: Spectral Clustering-Guided Co-Expression Network Analysis Without Scale-Free Constraints for Multi-Omic Data
>> https://www.biorxiv.org/content/10.1101/2025.04.27.650628v1
SGCRNA is a novel method for the analysis of co-expression networks, grounded in correlation and linear relationships, and independent of specific network topologies.
SCRNA employs spectral clustering. The complexity of calculating the eigenvectors of a square matrix of order is generally O(n3). It utilises an approximate method based on the Krylov subspace, reducing the complexity to O(k*n2), where k is the number of eigenvectors.

□ Tile-X: A vertex reordering approach for scalable long read assembly
>> https://www.biorxiv.org/content/10.1101/2025.04.21.649853v1
Tile-X, a novel graph-theoretic vertex reordering-centric approach to compute long read assemblies. The main idea of the approach is to efficiently compute an overlap graph first, use the overlap graph to (re)order the reads, and use that ordering to generate a parallel partitioned assembly.
Tile-RCM employs The Reverse Cuthill-McKee (RCM) ordering scheme is an efficient greedy heuristic that tries to minimize a measure of the graph's adjacency matrix bandwidth.
Tile-Metis is based on a graph partitioner, which uses a min-cut multi-level approach to generate a balanced partitioning of vertices (into a pre-specified number of partitions) and subsequently a traversal by each partition to generate its ordering.
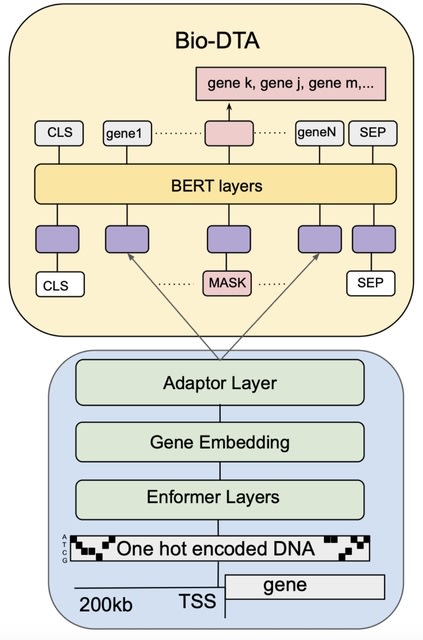
□ Bio-GTA: Multi-modal single-cell foundation models via dynamic token adaptation
>> https://www.biorxiv.org/content/10.1101/2025.04.17.649387v1
Extending the approach to all tokens in the input to allow their embeddings to flexibly encode additional information from a different modality that may change between data samples, which they call dynamic token adaptation (DTA).
Bio-DTA, a novel multi-modal model that learns from single-cell transcriptomes and DNA sequences jointly. It has learned dynamic co-regulation by assessing the impact of genetic changes to the DNA sequence of the transcription factor.
Bio-DTA combines a DNA language model with a single-cell foundation model. It receives a transcriptome of length 2,048 as an ordered sequence of gene names. Fixed token embeddings are replaced with projections of aggregated Enformer embeddings of each gene’s DNA sequence.

□ Detecting cell-level transcriptomic changes of Perturb-seq using Contrastive Fine-tuning of Single-Cell Foundation Models
>> https://www.biorxiv.org/content/10.1101/2025.04.17.649395v1
Pre-training a single-cell foundation model and fine-tune on a genome-scale perturbation dataset using a contrastive loss, which minimises the distance between cell embeddings from unperturbed cells while maximising the distance between perturbed and unperturbed cells.
The model is trained with a masked language modelling task where input tokens are randomly masked. The input to the model is a pair of transcriptomes: a perturbed and unperturbed cell form a dissimilar pair, or two unperturbed cells are a similar pair.

□ TWAVE: Generative prediction of causal gene sets responsible for complex traits
>> https://www.biorxiv.org/content/10.1101/2025.04.17.649405v1
Transcriptome-Wide conditional Variational auto-Encoder (TWAVE) uses a neural network encoder to embed high-dimensional gene expression profiles onto a low-dimensional latent space (Z), where data points can be classified and new representative points can be generated.
TWAVE employs a conditional VAE (a latent space explicitly trained to classify between baseline and variant). It draws from a probability distribution in the latent space associated with the trait phenotype label instead of drawing randomly from any state in the latent space.

□ S2-SPM: The Signed Two-Space Proximity Model for Learning Representations in Protein-Protein Interaction Networks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf204/8118643
The Signed Two Space Proximity Model (S2-SPM), the first archetypal-based signed network specifically tailored to model protein interactions. S2-SPM outperforms all compared baselines in terms of the tasks of sign and signed link prediction across three real-world PPI networks.
S2-SPM is supported by Gene Ontology-based enrichment analysis, clarifying the biological relevance of the identified archetypes. It assigns two latent vectors for each of the positive and negative interactions, projecting each protein to the two archetypal matrices/polytopes.
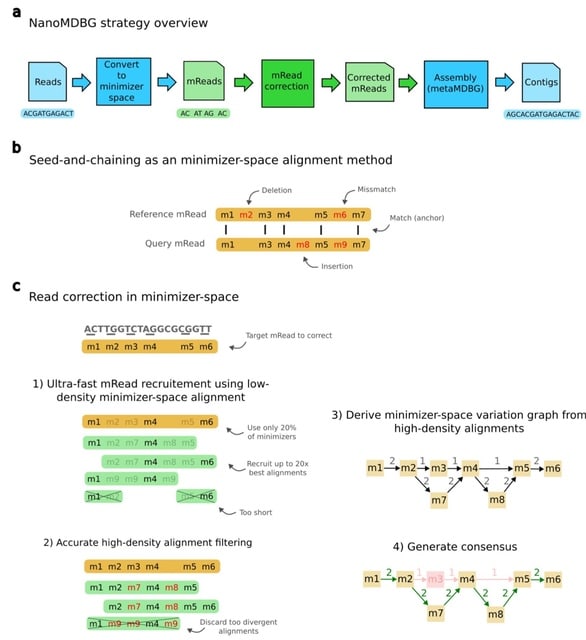
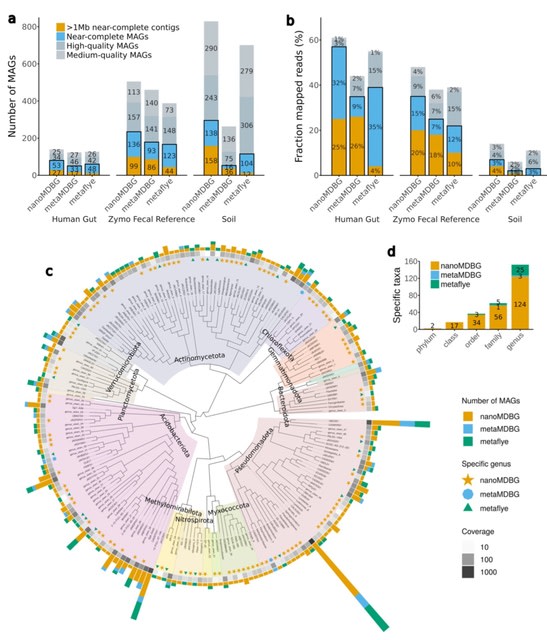
□ High-quality metagenome assembly from nanopore reads with nanoMDBG
>> https://www.biorxiv.org/content/10.1101/2025.04.22.649928v1
nanoMDBG, an evolution of the metaMDBG HiFi assembler, designed to support newer ONT sequencing data through a novel pre-processing step that performs fast and accurate error correction in minimizer-space.
Seed-and-chaining is usually a preliminary step before base-level alignment. Here, alignment operates entirely in minimizer-space without reverting to base-space, referred to as minimizer-space alignment. Sequence divergence is estimated directly from the result of chaining.
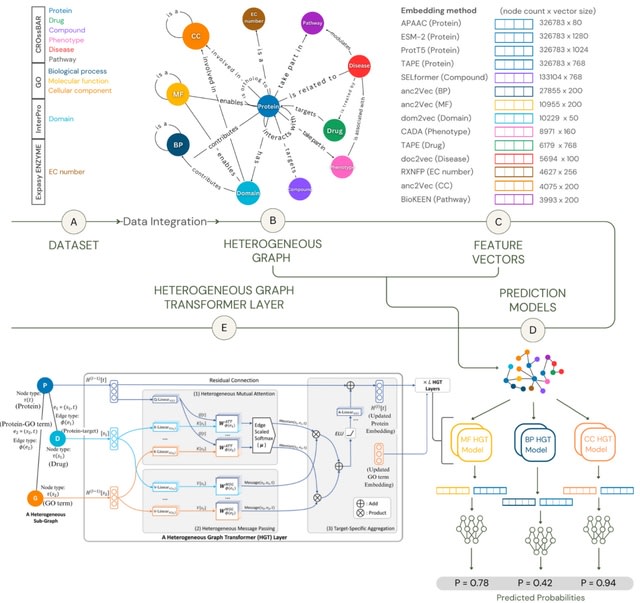
□ ProtHGT: Heterogeneous Graph Transformers for Automated Protein Function Prediction Using Biological Knowledge Graphs and Language Models
>> https://www.biorxiv.org/content/10.1101/2025.04.19.649272v1
ProtHGT is a heterogeneous graph transformer-based model that integrates diverse biological datasets into a unified framework using knowledge graphs. It leverages diverse biological entity types and highly representative protein language model embeddings at the input level.
ProtHGT effectively learns complex biological relationships, enabling accurate predictions across all Gene Ontology (GO) sub-ontologies.

□ CompleteBin: Dynamic Contrastive Learning with Pretrained Deep Language Model Enhances Metagenome Binning for Contigs
>> https://www.biorxiv.org/content/10.1101/2025.04.20.649691v1
CompleteBin trains a pretrained deep language model with dynamic contrastive learning and then clusters the contigs with their embeddings through the Leiden and FLSpp algorithms. It leverages both tetranucleotide frequencies and the sequence context of contigs as the input.
CompleteBin extracts the sequence context of contigs by sequence patch embedding, which is inspired by the patch embedding in the vision Transformer (ViT). CompleteBin pretrains half of the model layers with reference genomes and their taxonomic lineages.

□ m2ST: Dual Multi-Scale Graph Clustering for Spatially Resolved Transcriptomics
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf221/8119240
m2ST employs a multi-scale masked graph autoencoder to extract representations across different scales from spatial transcriptomic data. m2ST introduces a random masking mechanism for node features and employs a scaled cosine error as the loss function.
m2ST integrates scale-common and scale-specific information exploration into the clustering process, achieving more robust annotation performance. Shannon entropy is finally utilized to dynamically adjust the importance of different scales.

□ Ledidi: Programmatic design and editing of cis-regulatory elements
>> https://www.biorxiv.org/content/10.1101/2025.04.22.650035v1
Ledidi turns the design of edits into a continuous optimization problem using straight-through Gumbel-softmax reparameterization. A Gumbel-softmax distribution is defined through the addition of the log of the initial sequence plus a small epsilon and the learned weight matrix.
A discrete sequence -potentially containing edits- is generated and passed through a frozen model. Loss is calculated b/n the predicted outputs, and the straight-through estimator is used to pass the gradient through the discrete sequence and update the continuous weight matrix.

□ DESeq2-MultiBatch: Batch Correction for Multi-Factorial RNA-seq Experiments
>> https://www.biorxiv.org/content/10.1101/2025.04.20.649392v1
DESeq2-MultiBatch, a novel batch correction method that leverages DESeq2's internal model-based estimates to directly adjust raw count data, without relying on external correction tools or complex transformations.
DESeq2-MultiBatch preserves the integrity of log-fold changes b/n biological conditions and offers flexibility for handling complex experimental designs. It effectively accommodates interaction effects, even in scenarios characterized by imbalanced or highly confounded settings.

□ scStudio: A User-Friendly Web Application Empowering Non-Computational Users with Intuitive scRNA-seq Data Analysis
>> https://www.biorxiv.org/content/10.1101/2025.04.17.649161v1
scStudio is equipped with a suite of features designed to streamline data retrieval and analysis with both flexibility and ease, including automated dataset retrieval from the Gene Expression Omnibus (GEO).
The application supports all the essential steps required for scRNA-seq data analysis, including in-depth quality control, normalization, dimensionality reduction, clustering, differential expression and functional enrichment analysis.
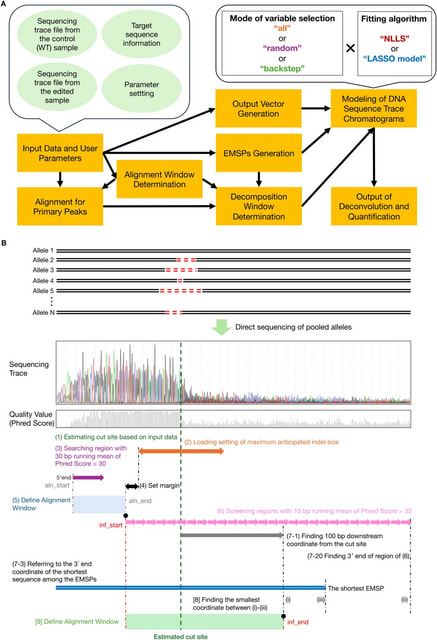
□ PtWAVE: a high-sensitive deconvolution software of sequencing trace for the detection of large indels in genome editing
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06139-8
PtWAVE (Progressive-type Wide-range Analysis of Varied Edits) constructs a more reliable mutation distribution by systematically selecting among various possible mutation patterns.
PtWAVE evaluates mutation distributions estimated using fitting algorithms and considers a lower Bayesian information criterion. It adjusts the combinations of estimated mutation sequence patterns (EMSPs) to estimate mutation distributions under reasonable conditions.

□ SCassist: An AI Based Workflow Assistant for Single-Cell Analysis
>> https://www.biorxiv.org/content/10.1101/2025.04.22.650107v2
SCassist consistently achieved high scores across groundedness, semantic similarity and expert human evaluation, indicating its ability to generate accurate and meaningful insights. SCassist generates metrics like summary statistics, quantile data, variance explained, and others.

□ On learning functions over biological sequence space: relating Gaussian process priors, regularization, and gauge fixing
>> https://www.biorxiv.org/content/10.1101/2025.04.26.650699v1
Establishing the relationship between regularized regression in overparameterized weight space and Gaussian process approaches that operate in "function space," i.e. the space of all real-valued functions on a finite set of sequences.
These connections arise naturally from the well-known link between L2-regularized and Bayesian linear regression, where the prior on weights is multivariate Gaussian. L2-regularization implicitly imposes a Bayesian prior on weight space.
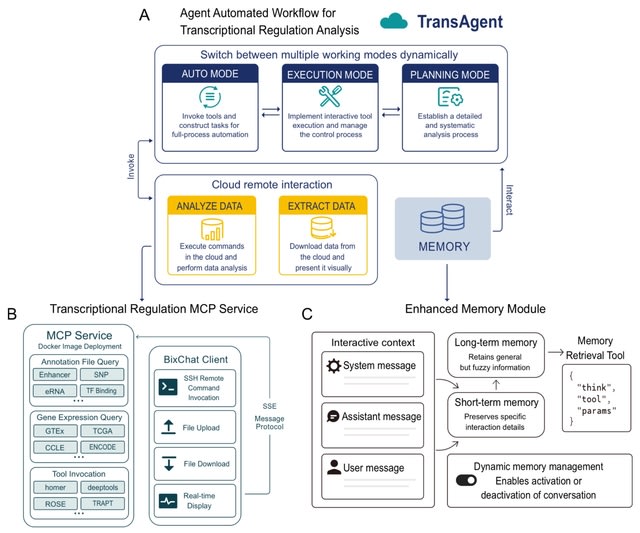
□ TransAgent: Dynamizing Transcriptional Regulation Analysis via Multi-omics-Aware AI Agent
>> https://www.biorxiv.org/content/10.1101/2025.04.27.650826v1
TransAgent—a LLM-driven software for transcriptional regulation analysis. Through intelligent task management and flexible tool calling, TransAgent effectively addresses these issues, enabling researchers to complete complex transcriptional regulation tasks more efficiently.
TransAgent captures user needs precisely through deep interaction, such as transcription factor activity prediction, binding prediction, epigenomic annotation, and gene expression analysis, and can generate detailed analysis workflows to ensure scientific reproducibility.
TransAgent flexibly calls various transcriptional regulation tools and provides real-time feedback during execution to ensure stable progress of the workflow. It completes the entire analysis process without manual intervention, significantly improving data processing efficiency.

□ DeepSAP: Improved RNA-Seq Alignment by Integrating Transcriptome Guidance with Transformer-Based Splice Junction Scoring
>> https://www.biorxiv.org/content/10.1101/2025.04.23.650072v1
DeepSAP (Deep Splice Alignment Program), a novel approach that integrates a new feature called Transcriptome-Guided Genomic Alignment (TGGA) in GSNAP, complemented by advanced deep learning techniques, such as fine-tuned transformer models.
The TGGA feature leverages a given transcriptome (such as those used by alignment-free methods) to allow for a relatively straightforward alignment of reads to known transcripts, while retaining the capacity for full genomic alignment to accommodate novel splice phenomena.
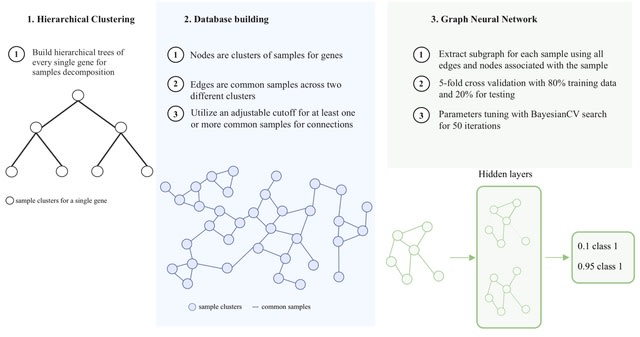
□ EGNF: EXPRESSION GRAPH NETWORK FRAMEWORK FOR BIOMARKER DISCOVERY
>> https://www.biorxiv.org/content/10.1101/2025.04.28.651033v1
Expression Graph Network Framework (EGNF), a cutting-edge graph-based approach that integrates graph neural networks (GNNs) with network-based feature engineering to enhance predictive biomarker identification.
EGNF constructs biologically informed networks by combining gene expression data and clinical attributes within a graph database, utilizing hierarchical clustering to generate dynamic, patient-specific representations of molecular interactions.
EGNF employs a bottom-up one-dimensional hierarchical clustering, using Euclidean distance as the similarity metric. Clusters are merged according to median linkage criteria, which calculates inter-cluster distances based on the median of pairwise distances b/n cluster centroids.


□ ParaHAT: Fast noisy long read alignment with multi-level parallelism
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06129-w
ParaHAT utilizes multi-level parallelism to accelerate alignment. ParaHAT focuses on parallel acceleration of rHAT without changing the original results. It optimizes the DP formula, eliminating intra-loop dependency, and further accelerating this process with vector-level parallelism.
ParaHAT proposes a general parallel alignment framework that accelerates the process by fully utilizing vector-level, thread-level, and process-level parallelism within a single node, and extends the algorithm across multiple computing nodes to further improve alignment speed.

□ STABIX: Summary statistic-based GWAS indexing and compression
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf264/8124076
STABIX, a compress and index method that improves upon bgzip compression ratios with an ensemble codec compression approach and improves upon Tabix queries with the addition of a summary-statistic-based index.
STABIX generates a genomic index which stores block, genomic, and file information necessary to reconstruct individual blocks.










※コメント投稿者のブログIDはブログ作成者のみに通知されます