









宇宙は荘厳である。
しかし荘厳な表面の下には何も無い。
愛も憎しみも、光も闇も。

□ GRIDSS, PURPLE, LINX: Unscrambling the tumor genome via integrated analysis of structural variation and copy number
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/25/781013.full.pdf
the combination of using GRIDSS for somatic structural variant calling and PURPLE for somatic copy number alteration calling allows highly sensitive, precise and consistent CN and SV determination, as well as providing novel insights for regions of complex local topology.
LINX, an interpretation tool, leverages the integrated structural variant and copy number calling to cluster individual structural variants into higher order events and chains them together to predict local derivative chromosome structure.


□ Autoencoder-based cluster ensembles for single-cell RNA-seq data analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/19/773903.full.pdf
An autoencoder-based cluster ensemble framework in which take random subspace projections, then compress each random projection to a low-dimensional space using an autoencoder artificial neural network, and apply ensemble clustering all encoded datasets for generating clusters.
The proposed framework of cluster ensemble via autoencoder-based dimension-reduction and its application to scRNA-seq is a principled approach and the first of its kind.

□ Dynamics of genetic code evolution: The emergence of universality
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/24/779959.full.pdf
The dynamics from the coevolution model along with the additional communication incorporated by the iterative discrete time algorithm governs the evolution of genetic code states (genetic code configurations) of which there is a finite number.
Consider this algorithm as if it were a system out of equilibrium for which there is the emergence of an attractor solution in the space of genetic code mappings.
The algorithm of Vetsigian provides a solution that is both optimal and universal. by allowing specific parameters to vary with time, the algorithm converges much faster to a universal solution. Automorphisms of the genetic code arising.

□ Vision: Functional interpretation of single cell similarity maps
>> https://www.nature.com/articles/s41467-019-12235-0
Vision operates directly on the manifold of cell-cell similarity and employs a flexible annotation approach that can operate either with or without preconceived stratification of the cells into groups or along a continuum.
the use of Vision within three different pipelines consisting of stratification free analysis where similarity between cells is based on either PCA or scVI, and stratification-based analysis where cells are organized along a developmental pseudo-time course.

□ f-VICE: Chromatin information content landscapes inform transcription factor and DNA interactions
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/23/777532.full.pdf
An information theory algorithms to measure signatures of TF-chromatin interactions encoded in patterns of the accessible genome, which they call chromatin information enrichment.
calculating chromatin information enrichment for hundreds of TF motifs across human tissues and find significant associations with TF-DNA residence times and specific DNA binding domains.
The extent of organization of data in the V-plot can be quantified using Shannon’s entropy, and detecting clusters of fragments distributed periodically in a “V” pattern indicating nucleosome phasing.

□ A multivariate phylogenetic comparative method incorporating a flexible function between discrete and continuous traits
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/20/776617.full.pdf
The multivariate approximate Bayesian computation - phylogenetic comparative method (ABC-PCM) allows the user to flexibly model an underlying latent evolutionary function between continuous and discrete traits.
Despite the fact that this analyses focused on a simple case in which a continuous trait affects the state of a discrete trait, other causational patterns can be dealt with by a flexible setting of an evolutionary simulation.
This framework can also be extended to model more complex evolutionary trajectories, such as asymmetric transitions between states and/or more than two states of discrete traits with different transition.

□ Chaos control in the fractional order logistic map via impulses
>> https://arxiv.org/pdf/1909.07110.pdf
The impulsive control, previously used in integer order continuous and discrete systems, is obtained by perturbing periodically (every ∆ steps) the state variable with a constant impulse: xn+1 ← (1+γ)xn+1.
If, for a chosen ∆, the control algorithm is applied for a γ value which generates in the bifurcation diagram versus γ a chaotic behavior, regular motions can be obtained. It is proved that the impulsed orbits remain bounded.
the control of chaos, or control of chaotic systems, is the boundary field between control theory and dynamical systems theory studying when and how it is possible to control systems exhibiting irregular, chaotic behavior.
□ Theory of high-dimensional outliers
>> https://arxiv.org/pdf/1909.02139v1.pdf
a new notion of high dimensional outliers that embraces various types and provides deep insights into understanding the behavior of these outliers based on several asymptotic regimes.
Geometrical properties of high dimensional outliers reveals an interesting transition phenomenon of outliers from near the surface of a high dimensional sphere to being distant from the sphere.


□ Chaotic synchronization induced by external noise in coupled limit cycle oscillators
>> https://arxiv.org/pdf/1909.08805.pdf
A solvable model of noise effects on globally coupled limit cycle oscillators. The averaged motion equation of the system with infinitely coupled oscillators is derived without any approximation through an analysis based on the nonlinear Fokker–Planck equation.
the occurrence of chaotic behavior in the order parameter system owing to the external noise, which is nonchaotic in the deterministic limit, would correspond to NICS.
To capture the dependence of attractor types on the Langevin noise intensity, the Lyapunov exponents for the system were numerically estimated, and could arbitrarily design the shape of the coupling function, which may yield a rich variety of dynamical behavior.
□ Induction of hierarchy and time through one-dimensional probability space with certain topologies
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/24/780882.full.pdf
The investment in adaptation in the higher order hierarchies diminishes chaotic behavior in the hierarchies.
Utilizing a Clifford algebra, a congruent zeta function, and a Weierstraß ℘ function in conjunction with a type VI Painlev ́e equation, the induction of hierarchy and time through one-dimensional probability space with certain topologies.

□ GripDL: Predicting gene regulatory interactions based on spatial gene expression data and deep learning
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007324
GripDL (Gene regulatory interaction prediction via Deep Learning), which incorporates high-confidence TF-gene regulation knowledge. GripDL uses a ResNet pretrained on the ImageNet database as the initial model, which is actually a transfer learning strategy.
GripDL achieves significant improvement on the predicting accuracy compared to unsupervised reconstructing methods, suggesting the successful transfer of the TF-target regulation knowledge to the recognition of spatial patterns for identifying new regulatory interactions.
□ Multi-Cell ECM compaction is predictable via superposition of nonlinear cell dynamics linearized in augmented state space
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006798
This method recasts the original nonlinear dynamics in an augmented space where the system behaves more linearly.
The collective ECM compaction by multiple cells is predicted through superposition of individual cells’ contributions in latent variable space.
□ centroFlye: Assembling Centromeres with Long Error-Prone Reads
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/16/772103.full.pdf
the centroFlye algorithm for centromere assembly using long error-prone reads, apply it for assembling the human X centromere, and use the constructed assembly to gain insights into centromere evolution.
This analysis reveals putative breakpoints in the previous manual reconstruction of the human X centromere and opens a possibility to automatically close the remaining multi-megabase gaps in the reference human genome.
□ Assexon: Assembling Exon Using Gene Capture Data:
>> https://journals.sagepub.com/doi/10.1177/1176934319874792
Assexon: a streamlined pipeline that de novo assembles targeted exons and their flanking sequences from raw reads.
Assexon accurately assembled more than 3400 to 3800 (20%-28%) loci than PHYLUCE and more than 1900 to 2300 (8%-14%) loci than HybPiper across different levels of phylogenetic divergence.
□ RamDA-seq: Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs
>> https://www.nature.com/articles/s41467-018-02866-0
random displacement amplification sequencing (RamDA-seq), the first full-length total RNA-sequencing method for single cells.
The sensitivity and full-length transcript coverage of RamDA-seq were achieved using RT-RamDA and not-so-random primers (NSRs).
RamDA-seq profiles recursive splicing in >300-kb introns. RamDA-seq also detects enhancer RNAs and their cell type-specific activity in single cells.
□ SCSsim: an integrated tool for simulating single-cell genome sequencing data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz713/5570983
SCSsim first constructs the genome sequence of single cell by mimicking a complement of genomic variations under user-controlled manner.
Second, the WGA procedure is implemented as dividing the single cell genome into variable-size fragments and amplifying the fragments by emulating MALBAC technique.
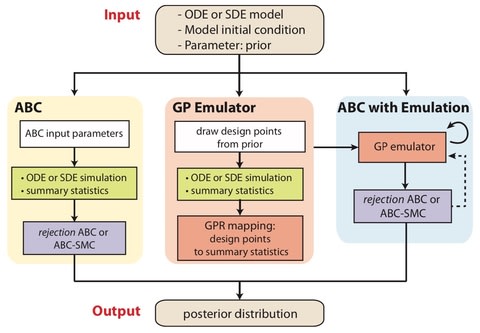
□ GpABC: a Julia package for approximate Bayesian computation with Gaussian process emulation
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/18/769299.full.pdf
an extensible Julia package, GpABC, which implements rejection ABC and ABC-SMC with GP emulation for parameter and model inference in deterministic and stochastic models.
Biochemical reactions are stochastic in nature, and the distribution of stochastic simulation trajectories is generally non-Gaussian.
To meet the Gaussian noise assumption of a GP and to consider computational efficiency, employ the linear noise approximation (LNA), a first-order expansion of the stochastic differential equation.
Training the GP has computational complexity O(n3), and emulating the model has complexity of O(bn), assuming batch size b.
□ ascend: R package for analysis of single-cell RNA-seq data
>> https://academic.oup.com/gigascience/article/8/8/giz087/5554286
ascend’s streamlined workflow includes filtering, normalization, dimension reduction, clustering, differential expression, and visualization.
ascend optimizes parallelization and algorithms for improving speed of each analysis step, and implements Clustering by Optimal REsolution (CORE) for unsupervised, robust hierarchical clustering.
□ scHiCTools: a computational toolbox for analyzing single cell Hi-C data
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/18/769513.full.pdf
scHiCTools includes three smoothing approaches. Linear convolution is based on a 2D filters (a.k.a., convolution kernels) with equal values in every position, which can be viewed as smoothing over nearby bins in Hi-C contact maps.
scHiCTools implements a faster version of HiCRep, together with another Hi-C similarity measure named Selfish, and a new inner product approach which provides a more efficient way of embedding scHi-C data. All of the three approaches have O(n) computational complexity.

□ scSEGIndex: Evaluating stably expressed genes in single cells
>> https://academic.oup.com/gigascience/article/8/9/giz106/5570567
stably expressed genes (SEGs) identified from single cells based on their stability indices are considerably more stable than HKGs defined previously from cell populations across diverse biological systems.
To assess the expression stability of each gene list in various cell types and biological systems, the k-means algorithm was used to cluster each scRNA-seq dataset to its predefined number of clusters,
and an array of evaluation metrics were applied to compute the concordance with respect to the predefined (“gold standard”) class labels. Evaluation metrics include the ARI, Purity, FM, and the Jaccard index.
□ Controlled Self-Assembly of λ-DNA Networks with the Synergistic Effect of DC Electric Field
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/19/774901.full.pdf
a series of large-scale and morphologically controlled self-assembled λ-DNA networks were successfully fabricated by the synergistic effect of DC electric field.
DNA molecules were obviously stretched in both horizontal and vertical electric fields at low DNA concentrations.

□ BITACORA: A comprehensive tool for the identification and annotation of gene families in genome assemblies
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/19/593889.full.pdf
BITACORA, a bioinformatics solution that integrates sequence similarity search tools and Perl scripts to facilitate both the curation of these inaccurate annotations and the identification of previously undetected gene family copies directly from DNA sequences.
The output of BITACORA can be used as a baseline for manual annotation in genomic annotation editors, used as evidence in automatic annotation tools to improve gene family model predictions, or to directly perform downstream analysis.

□ UMI-VarCal: a new UMI-based variant caller that efficiently improves low-frequency variant detection in paired-end sequencing NGS libraries
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/19/775817.full.pdf
UMI-VarCal, a somatic single nucleotide variant and indel caller for UMI-based targeted paired-end sequencing protocols.
a Poisson statistical test is applied at every position to determine if the frequency of the variant is significantly higher than the background error noise.
UMI-VarCal stands out from the crowd by being one of the few variant callers that don’t rely on SAMtools to do their pileup. UMI-VarCal is faster than both raw-reads-based and UMI-based variant callers.
□ ConnectedReads: machine-learning optimized long-range genome analysis workflow for next-generation sequencing
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/20/776807.full.pdf
an efficient and effective whole-read assembly workflow with unsupervised graph mining algorithms on an Apache Spark large-scale data processing platform called ConnectedReads.
By fully utilizing short-read data information, ConnectedReads is able to generate haplotype-resolved contigs and then streamline downstream pipelines to provide higher-resolution SV discovery than that provided by other methods, especially in N-gap regions.

□ Gene capture by transposable elements leads to epigenetic conflict
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/20/777037.full.pdf
Syntelog genes appeared to maintain function, consistent with moderation of the epigenetic response for important genes before reaching a deleterious threshold, while transposed genes bore the signature of silencing and potential pseudogenization.
Intriguingly, transposed genes were overrepresented among donor genes, suggesting a link between capture and gene movement.

□ Perfect quantum state transfer on diamond fractal graphs
>> https://arxiv.org/pdf/1909.08668v1.pdf
the analysis of perfect quantum state transfer beyond one dimensional spin chains to show that it can be achieved and designed on a large class of fractal structures, known as diamond fractals, which have a wide range of Hausdorff and spectral dimensions.
The resulting systems are spin networks combining Dyson hierarchical model structure and transport properties of one dimensional chains with transverse permutation symmetries of varying order.
This approach allows to consider other transport phenomena involving linear and nonlinear, classical and quantum waves on certain graphs, quantum graphs, and fractals.

□ ARMADA: A statistical methodology to select covariates in high-dimensional data under dependence
>> https://arxiv.org/pdf/1909.05481v1.pdf
The methodology successively intertwines the clustering of covariates, decorrelation of covariates using Factor Latent Analysis.
ARMADA takes into account the structure of correlation by clusters of covariates, and applying a ”decorrelation” between the covariates inside each cluster.

□ PMMLogit: High-dimensional Bayesian phenotype classification and model selection using genomic predictors
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/23/778472.full.pdf
a Bayesian hierarchical model termed ‘PMMLogit’ for classification and model selection in high-dimensional settings with binary phenotypes as outcomes.
Previously developed approaches in this setting have relied on the Laplace approximation or the Metropolis-Hastings algorithm.
combine a Polya-Gamma based data augmentation strategy and use recent results on Markov chain Monte-Carlo (MCMC) techniques to develop an efficient and exact sampling strategy for the posterior computation.

□ DAVI: Deep Learning Based Tool for Alignment and Single Nucleotide Variant identification
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/23/778647.full.pdf
a novel deep neural network based tool "DAVI" (Deep Alignment and Variant Identification) consists of models for both global and local alignment and for Variant Calling.
DAVI uses CNN like DeepVariant but instead of using pileup images with inception-v2 architecture, and use Position Specific Frequency Matrix(PSFM) to identify possible variant sites.
□ cuteSV: Long Read based Human Genomic Structural Variation Detection
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/24/780700.full.pdf
cuteSV, a sensitive, fast and lightweight SV detection approach. cuteSV uses tailored methods to comprehensively collect various types of SV signatures, and a clustering-and-refinement method to implement a stepwise SV detection.
cuteSV employs a stepwise refinement clustering algorithm to process the comprehensive signatures from inter- and intra-alignment, construct and screen all possible alleles thus completes high-quality SV calling.
□ CNVmap: a method and software to detect copy number variants from linkage mapping data
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/24/778753.full.pdf
an original linkage-based method to detect CNVs from genotype data of mapping populations.
This software based on this method makes it possible to perform fully automatic mining of segregation data to extract a list of high confidence CNVs, including the detailed type of event and the genomic location(s) of the initially unknown locus or loci.
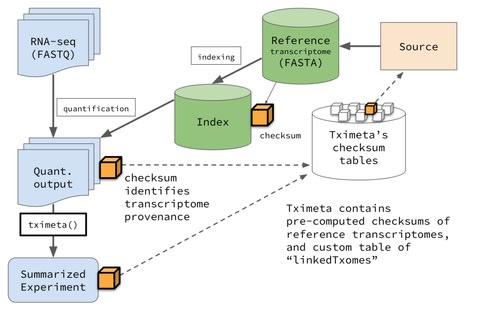
□ Tximeta: reference sequence checksums for provenance identification in RNA-seq
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/25/777888.full.pdf
a solution in the form of an R/Bioconductor package tximeta that performs numerous annotation and metadata gathering tasks automatically on behalf of users during the import of transcript quantification files.
The computational paradigm of automatically adding annotation metadata based on reference sequence checksums can greatly facilitate genomic workflows, by helping to reduce overhead during bioinformatic analyses, preventing costly bioinformatic mistakes, and promoting computational reproducibility.

□ EAGLE: an algorithm that utilizes a small number of genomic features to predict tissue/cell type-specific enhancer-gene interaction
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/25/781427.full.pdf
EAGLE, an enhancer and gene learning ensemble method for identification of Enhancer-Gene (EG) interactions measured by prediction probabilities.
EAGLE used only six features derived from the genomic features of enhancers and gene expression datasets, and displayed a better performance in the 10-fold cross-validation and cross-sample test.

□ It's about time: Analysing an alternative approach for reductionist modelling of linear pathways in systems biology
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/25/781708.full.pdf
a computational investigation of linear pathway models that contain fewer pathway steps than the system they are designed to emulate.
assuming a fixed rate of information propagation along a pathway of dynamic length. This leads to a three-parameter model which can recapture the dynamics of arbitrary linear pathways with high fidelity.
□ A Coding Framework for Improving Transparency in Decision Modeling
>> https://link.springer.com/article/10.1007/s40273-019-00837-x
The proposed framework consists of a conceptual, modular structure and coding recommendations for the implementation of model-based decision analyses in R.
The analysis component is the application of the fully developed decision model to answer the policy or the research question of interest, assess decision uncertainty, and/or to determine the value of future research through value of information (VOI) analysis.

□ Complex genetic and epigenetic regulation deviates gene expression from a unifying global transcriptional program
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007353
an integrated framework of resource allocation that a rich structure of deviations from it exists and that by characterizing these deviations we can fully appreciate large-scale expression change.
The balance between regulatory strategies ultimately modulates the action of the general transcription machinery and therefore limits the possibility of establishing a unifying program of expression change at a genomic scale.
□ Single Cell Explorer: collaboration-driven tools to leverage large-scale single cell RNA-seq data
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-6053-y
Single Cell Explorer is a Python-based web server application to enable computational and experimental scientists to iteratively and collaboratively annotate cell expression phenotypes within a user-friendly and visually appealing platform.
Data processing and analytic workflows can be integrated into the system using Jupyter notebooks. This step includes read mapping alignment, gene quantitation, and quality control employing Cell Ranger v3.0 to process Chromium single-cell RNA-seq FASTQ data.
□ GeneRax: A tool for species tree-aware maximum likelihood based gene tree inference under gene duplication, transfer, and loss
>> https://www.biorxiv.org/content/biorxiv/early/2019/09/26/779066.full.pdf
GeneRax is the fastest among all tested methods when starting from aligned sequences, and that it infers trees with the highest likelihood score.
compared to competing tools, on simulated data GeneRax infers trees that are the closest to the true tree in 90% of the simulations in terms relative Robinson-Foulds distance.










※コメント投稿者のブログIDはブログ作成者のみに通知されます