









□ affenfaustHH:
"Das eigene Ich" @bjoern_holzweg "Clubber Lang" 120x120 cm, Bleistift & Aquarell auf Papier,2015


□ NEMix: Single-cell Nested Effects Models for Probabilistic Pathway Stimulation:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004078
Structure learning is performed using a greedy heuristic to find an optimal network. Similar to the NEM procedure described in, edges are incrementally added if the likelihood is increased.
□ Halvade: scalable sequence analysis with MapReduce: parallel on a multi-node and multi-core compute infrastructure
>> http://bioinformatics.oxfordjournals.org/content/early/2015/04/15/bioinformatics.btv179.full.pdf
Using a 15-node computer cluster with 360 CPU cores in total, Halvade processes the NA12878 dataset human, 100 bp paired-end reads, 50× coverage in <3 h with very high parallel efficiency. During the map phase, read alignment is performed by BWA; both BWA-mem and BWA-aln with BWA-sampe on a single 24-core node with three parallel tasks, Halvade already attains a speedup of 2.5 compared with a multithreaded execution of the same tools in a single task. A second key factor in attaining a high efficiency is the choice of appropriate task sizes.
<br />
□ Belgian Team Reports Protocol to Increase NimbleGen SeqCap EZ Multiplexing Capacity:
>> https://www.genomeweb.com/microarrays-multiplexing/belgian-team-reports-protocol-increase-nimblegen-seqcap-ez-
□ The State of Probabilistic Programming:
>> https://moalquraishi.wordpress.com/2015/03/29/the-state-of-probabilistic-
Probabilistic Programming for Advanced Machine Learning has resulted in a great infusion of energy and funding. Deep learning has momentarily sucked the air out of the machine learning room and the slowness and generally poor performance of sampling-based approaches hasn’t helped the cause of probabilistic programming either.

□ Deep Learning for Bioinformatics:
>> http://etrigg.com/event/deep-learning-for-bioinformatics,1013297/
PconsC2 uses a deep learning approach to identify protein-like contact patterns to improve contact predictions. the method described here is different from earlier deep-learning approaches, that implementation is based on a feed-forward stack of random forests learners and not layers of Neural Networks

GQT enables queries based on sample genotypes and phenotypes.
□ Genotype Query Tools (GQT): Efficient compression and analysis of large genetic variation datasets
>> http://biorxiv.org/content/biorxiv/early/2015/04/20/018259.full.pdf
a new indexing strategy and powerful toolset that enables interactive analyses based on genotypes, phenotypes and sample relationships. "variant-centric" indexing strategies can significantly expand the capabilities of population-scale analyses by providing interactive-speed queries to data sets with millions of individuals.
□ Artificial Intelligence has crushed all human records in 2048. Here’s how the AI pulled it off.
>> http://www.randalolson.com/2015/04/27/artificial-intelligence-has-crushed-all-human-records-in-2048-heres-how-the-ai-pulled-it-off/ …
□ 2048-exome: http://stephenturner.us/2048-exome/
□ scikit-neuralnetwork: Deep neural network implementation without the learning cliff!
>> https://github.com/aigamedev/scikit-neuralnetwork
In particular, this model has a single hidden layer with 300 hidden units of type Rectified Linear (ReLU) and trained with the same data with validation and monitoring disabled.
Convolution("Rectifier", channels=8, kernel_shape=(3,3)),
Layer("Softmax")],
learning_rate=0.02,
□ An overview of the BIOASQ large-scale biomedical semantic indexing and question answering competition:
>> http://www.biomedcentral.com/1471-2105/16/138/abstract …
□ Pydna: a simulation and documentation tool for DNA assembly strategies using python:
>> http://www.biomedcentral.com/1471-2105/16/142/abstract …
□ Fine-mapping cellular QTLs with RASQUAL and ATAC-seq:
>> http://www.biorxiv.org/content/early/2015/04/30/018788
association mapping that integrates genetic effects and robust modelling of biases in NGS data within a single, probabilistic framework.
□ Exploring Free Energy Landscapes of Large Conformational Changes: Molecular Dynamics with Excited Normal Modes:
>> http://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00003 …
the MDeNM method consists of multiple-replica short MD simulations described by a given subset of low-frequency NMs are kinetically excited. This is achieved by adding additional atomic velocities along several randomly determined linear combinations of NM vectors,
□ GFVO: the Genomic Feature and Variation Ontology:
>> https://peerj.com/articles/933/
GFVO is modeled in OWL and made available in RDF/XML format. The ontology’s expressiveness is within SRIQ with datatype properties, which is a Description Logic fragment
□ The Biology Of Genomes May 5 - 9, 2015 organized at and by Cold Spring Harbor Lab, NY #bog15

□ Kallisto, a new ultra fast RNA-seq quantitation method:
>> http://nextgenseek.com/2015/05/kallisto-a-new-ultra-fast-rna-seq-quantitation-method/
The Kallisto github page also shows the use of an EM algorithm on the pseudoalignments to resolve read origin ambiguities in reads aligning. On benchmarks with standard data, kallisto can quantify 30 million human reads in less than 3 minutes on a Mac using only the read sequences.
the TopHat + Cufflinks combo took something along the lines of a half a day per sample, using multithreading over 12 cores for each sample. The Salmon quantification took about 15 minutes per sample, Kallisto took about 10 minutes per sample and does not do any multithreading.
□ DrKatHolt:
ANDI https://github.com/evolbioinf/andi/… is really awesome... Just made a tree of 100 genomes from 2 related spp while sitting at #ABPHM15. ~60 secs.
andi uses an algorithm for rapidly computing the evolutionary distances between closely related genomes. distance measure is based on ungapped local alignments that we anchor through pairs of maximal unique matches of a minimum length. These exact matches using enhanced suffix arrays and this implementation requires approximately only 1 s and 45 MB RAM/Mbase analysed.
□ jdidion:
Where do species-specific active regulatory elements come from? Ans: neo-functionalization of ancestral sequence. @CamLBerthelot #BOG15
□ Dimensionality reduction for zero-inflated single cell gene expression analysis:
>> http://biorxiv.org/content/early/2015/05/08/019141
The fundamental empirical observation that underlies the zero-inflation model in ZIFA is that the dropout rate. if the mean level of non-zero expression, this dropout relationship can be approximately modelled with a parametric form p0 = exp(-λμ^2)
test the relative performance of ZIFA against Principal Components Analysis (PCA), Probabilistic PCA (PPCA), Factor Aanalysis and, where appropriate, non-linear techniques including Stochastic Neighbour Embedding (t-SNE), Isomap, and Multidimensional Scaling (MDS).
□ A Vision for Ubiquitous Sequencing:
>> http://biorxiv.org/content/biorxiv/early/2015/05/07/019018.full.pdf
Bringing sequencing sensors to the masses also has the advantage of crowd-sourcing DNA or RNA signatures from various sources. Most sequence analysis algorithms are devised as offline programs that assume that all input data is available at the beginning of the run and that files are read and written at each stage.
□ A multi-view genomic data simulator: ordinary differential equations w/ objective of benchmarking multi-view learning
>> http://www.biomedcentral.com/content/pdf/s12859-015-0577-1.pdf
Due to the high-dimensional nature of OMICs data, effective modelling for inference or prediction in bioinformatics cannot be performed without an initial phase of feature selection. MVBioDataSim shows that the generated datasets are well mimicking the behaviour of real data, for popular network reconstruction methods are able to selectively identify existing interactions.
□ BrAD-seq: Breath Adapter Directional sequencing: a streamlined, ultra-simple and fast library preparation protocol:
>> http://journal.frontiersin.org/article/10.3389/fpls.2015.00366/abstract …

□ LDetect: Approximately independent linkage disequilibrium blocks in human populations:
>> http://biorxiv.org/content/biorxiv/early/2015/06/01/020255.full.pdf
Calculate the n×n covariance matrix C for all pairs of loci using the shrinkage estimator of C, Convert the covariance matrix to n × n matrix of squared Pearson product- moment correlation coefficients P, Convert the matrix P = (ei,j) to a (2n - 1)-dimensional vector V = (vk)
In reality, matrix P turns out to be sparse, approximately banded, and approx- imately block-diagonal, with sporadically overlapping blocks
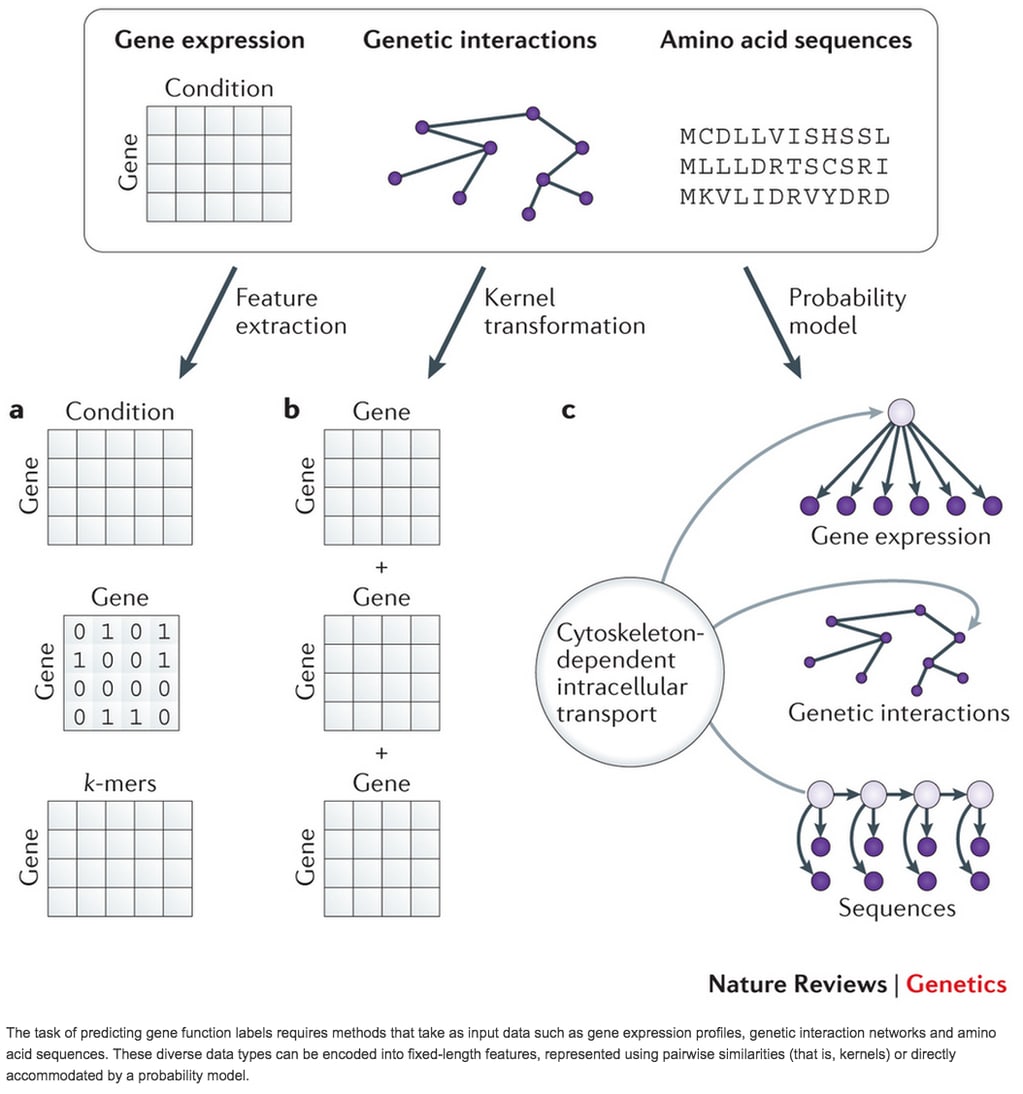
□ Machine learning applications in genetics and genomics
>> http://www.nature.com/nrg/journal/vaop/ncurrent/full/nrg3920.html
□ Sequencing ultra-long DNA molecules with the Oxford Nanopore MinION:
>> http://biorxiv.org/content/biorxiv/early/2015/05/13/019281.full.pdf
The protocol for preparing a MinION sequencing library is still evolving, but currently includes shearing genomic DNA using a Covaris g-TUBE, an optional “PreCR” step to repair damaged DNA, end-repair, dA-tailing, adapter ligation, and His-bead purification.
□ De Bruijn Graphs and pynteractive: Being able to debug graphically gene assembly
>> https://vimeo.com/128206058
□ rafalab:
Completely confounded designs ruin experiments. Combat/SVA/RUV won't save you. Consider randomization.
Is it species or is it batch? They are confounded, so we can't know:
>> http://simplystatistics.org/2015/05/20/is-it-species-or-is-it-batch-they-are-confounded-so-we-cant-know/ …
□ dgmacarthur:
Users should not accept "black box" approaches from bioinformatics companies:
>> http://www.nature.com/news/no-more-hidden-solutions-in-bioinformatics-1.17587
□ tonyhammond:
New http://www.nature.com/ontologies/ . Grown core by 50%, two more domain models, MeSH links, better navigation. #linkeddata

the NPG Core Ontology: the RDF(S) and OWL foundational models and to the SKOS model.

Side and top views of geometry optimized TBLG with the twist angles θ of (a) 29.4° and (b) 8.26°.
□ Large-scale real-space density-functional calculations: Moiré-induced electron localization in graphene:
>> http://scitation.aip.org/content/aip/journal/jap/117/11/10.1063/1.4913837 …
the introduction of suitable mathematical schemes, allows us to carry out density-functional calculations for the Si nanowire system containing 107 292 atoms using 82 944 nodes of the K computer with 51.7% efficiency.
□ Determining Exon Connectivity in Complex mRNAs by Nanopore Sequencing:
>> http://biorxiv.org/content/biorxiv/early/2015/05/22/019752.full.pdf …
MinION can be used to easily determine the connectivity of exons in a single transcript, even for the most complicated alternatively spliced genes. nanopore sequencing of whole transcriptomes, rather than targeted genes, will be a rapid and powerful approach for characterizing isoforms, especially with improvements in the throughput and accuracy of the technology, and the simplification and/or elimination of the time-consuming library preparations.
□ Pachter’s P-value Prize: $100/P: Bits of DNA
>> https://liorpachter.wordpress.com/2015/05/26/pachters-p-value-prize/ …
to the person who can best justify a reasonable null model, together with a p-value (p) for the phrase “Strikingly, 95% of cases of accelerated evolution involve only one member of a gene pair”
□ Long-term survival of duplicate genes despite absence of subfunctionalized expression:
>> http://biorxiv.org/content/biorxiv/early/2015/05/10/019166.full.pdf
Minor genes do show clear evidence of functional constraint: 97% of minor genes have dN /dS <1, a hallmark of protein-coding constraint. Subsequently, the relative expression levels of the two genes evolve as a random walk, so slowly due to constraint on combined expression. If expression happens to become asymmetric, this reduces functional constraint on the minor gene.
<br />
□ Sequencing, Finishing, and Analysis in the Future Meeting 2015 sponsored by Los Alamos natl Lab in Santa Fe
>> http://www.lanl.gov/conferences/sequencing-finishing-analysis-future/index.php … #SFAF2015
The meeting focuses on laboratory methods and computational tools used to help sequence, assemble, and finish genomes, including new sequencing technologies, which promise high-throughput results by sequencing more base-pairs per run at longer read-lengths.
□ finchtalk:
#SFAF2015 WS X ten data: 1 genome ~0.5 TB (seqs, aligns, vars) deal with what to keep. 1500 samples/month = 600 TB. Wants to go BAM-free.
□ EDGE: Empowering the Development of Genomics Expertise:
>> https://edge.readthedocs.org/en/latest/
EDGE is a highly adaptable bioinformatics platform that allows users to address a wide range of use cases including assay validation and the characterization of novel biological threats , clinical samples, and complex environmental samples.
"apparently, only assembly and read mapping are two bioinformatics analysis needed"
□ crashfrog:
Power8 architecture: http://en.wikipedia.org/wiki/POWER8 might be the "missing middle" for high-perf bioinf btwn Hadoop and HPC
□ Large-scale Machine Learning for Metagenomics Sequence Classification:
>> http://arxiv.org/pdf/1505.06915v1.pdf
Tuning these models involves training a machine learning model on about 10^8 samples in 10^7 dimensions,
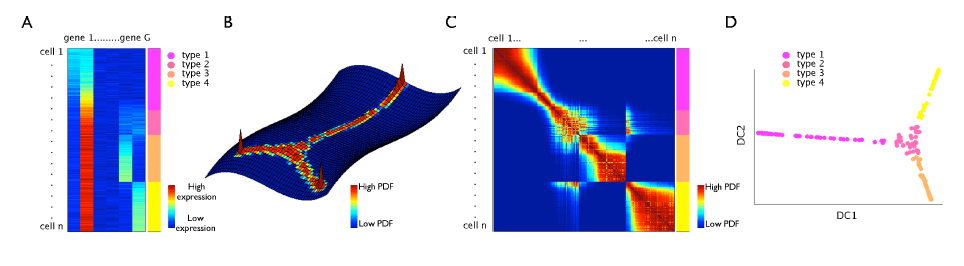
□ Diffusion maps for high-dimensional single-cell analysis of differentiation data:
>> http://bioinformatics.oxfordjournals.org/content/early/2015/05/21/bioinformatics.btv325.abstract
adapt this method to single-cell data by adequate choice of kernel width and inclusion of uncertainties or missing measurement values, which enables the establishment of a pseudo-temporal ordering of single cells in a high-dimensional gene expression space.
D_=diag(sum(H));
Hp=D_^(-1)*H;
[psi_nsort,En]=eig(Hp);
%[psi_nsort,En]=eigs(Hp); %to calculate only few largest
%eigenvalues/eigenvectors
□ Spacetime SQUIDs: Building an artificial spacetime in the lab to test quantum gravity & inflation:
>> http://fqxi.org/community/articles/display/201

□ How spacetime is built by quantum entanglement
>> http://www.eurekalert.org/pub_releases/2015-05/uot-hsi052715.php
The interface between quantum gravity and information science is becoming increasingly important for both fields. quantum entanglement generates the extra dimensions of the gravitational theory.

□ Markov Composer - Using machine learning and a Markov chain to compose music:
>> http://zx.rs/3/Markov-Composer---Using-machine-learning-and-a-Markov-chain-to-compose-music/ …










※コメント投稿者のブログIDはブログ作成者のみに通知されます