








 (Photo by Natan Vance)
(Photo by Natan Vance)The promise doesn't serve any purpose. Time manifests itself in the process of revealing immanence, and past and future compositions are always being tested at this moment.
『約束』は意味を為さない。時が顕現するのは内在性の露呈する過程であり、過去と未来のコンポジションは常に今この瞬間に試されている。

□ Wavefront Alignment Algorithm: Fast gap-affine pairwise alignment using the wavefront algorithm
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa777/5904262
WFA, the wavefront alignment algorithm - an exact gap-affine algorithm that takes advantage of homologous regions between the sequences to accelerate the alignment process.
As opposed to traditional dynamic programming algorithms that run in quadratic time, the WFA runs in time O(ns), proportional to the read length n and the alignment score s, using O(s2) memory.
the WFA runs 20-300x faster than other methods aligning short Illumina-like sequences, and 10-100x faster using long noisy reads like those produced by Oxford Nanopore Technologies.
Wavefront Alignment Algorithm can be easily vectorized using SIMD, by the automatic features of modern compilers, for different architectures, without the need to adapt the code, and naturally computes cells of the DP matrix by increasing score without introducing further complexities.
□ Heng Li @lh3lh3
WFA is a non-heuristic algorithm for doing Needleman-Wunsch alignment with affine gap penalty. Its time complexity is linear in the sequence divergence, making it much faster than other NW equivalent on similar sequences. A breakthrough. github.com/smarco/WFA

□ VeTra: a new trajectory inference tool based on RNA velocity
>> https://www.biorxiv.org/content/10.1101/2020.09.01.277095v1.full.pdf
VeTra reconstructs the pseudo-temporal orders of the cells based on the coordinate and the velocity vector of each cell in the low-dimensional space. Given velocity vectors, VeTra reconstructs a directed graph.
VeTra can cluster multifurcated trajectories. VeTra provides a flexible environment to obtain cell group, select trajectory of interest and enables pseudo-time analysis.

□ uLTRA: Accurate spliced alignment of long RNA sequencing reads
>> https://www.biorxiv.org/content/10.1101/2020.09.02.279208v1.full.pdf
uLTRA aligns long-reads to a genome using an exon annotation. uLTRA solves the algorithmic problem of chaining with overlaps to find alignments. To align reads, uLTRA first finds maximal exact matches (MEMs) between the reads and the parts and flanks using slaMEM.
uLTRA finds a collinear chain of MEMs covering as much of the read as possible. uLTRA aligns these segments together with all small exons using edlib. The collinear chaining solution of MAMs is used to produce the final alignment of the read to the genome.

□ DISSECT: DISentangle SharablE ConTent for Multimodal Integration and Crosswise-mapping
>> https://www.biorxiv.org/content/10.1101/2020.09.04.283234v1.full.pdf
a self-supervised deep learning-based approach that leverages unpaired data across two domains to learn crosswise mapping while disentangling mutual information content from distinct profiling modalities at single-cell resolution on a toy dataset.
DISSECT identies domain-specific information from sets of unpaired measurements in complementary data domains by considering a deep learning cross-domain autoencoder architecture designed to learn shared latent representations of data while enabling domain translation.

□ PRESCIENT: Generative modeling of single-cell population time series for inferring cell differentiation landscapes
>> https://www.biorxiv.org/content/10.1101/2020.08.26.269332v1.full.pdf
PRESCIENT (Potential eneRgy undErlying Single Cell gradIENTs), a generative modeling framework that learns an underlying differentiation landscape from single-cell time-series gene expression data.
PRESCIENT’s generative model framework provides insight into the process of differentiation and can simulate differentiation trajectories for arbitrary gene expression progenitor states.

□ Entropic Regression: How entropic regression beats the outliers problem in nonlinear system identification
>> https://aip.scitation.org/doi/full/10.1063/1.5133386
Entropic Regression, a nonlinear System Identification (SID) methods, whereby true model structures are identified based on an information-theoretic criterion describing relevance in terms of reducing information flow uncertainty vs not necessarily sparsity.
Entropic Regression combines entropy measures with an iterative optimization for nonlinear SID. Entropic Regression can be thought of as an information-theoretic extension of the orthogonal least squares regression or as a regression version of optimal causation entropy.

□ Limit cycles in models of circular gene networks regulated by negative feedback loops
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03598-z
The non-stationary dynamics in the models of circular gene networks with negative feedback loops is achieved by a high degree of non-linearity of the mechanism of the autorepressor influence on its own expression.
The models of these gene networks possess not only a high oscillatory potential but also the possibility of complex, chaotic dynamics formation, including the hyperchaotic one.
The main aim of the investigations is nonlocal analysis of some gene networks models represented by dynamical systems of kinetic type. the analysis of phase portraits of the higher-dimensional gene networks models with negative feedback loops, delayed arguments phenomena.
□ BayesSpace: the robust characterization of spatial gene expression architecture in tissue sections at increased resolution
>> https://www.biorxiv.org/content/10.1101/2020.09.04.283812v1.full.pdf
BayesSpace is the first spatial transcriptomics model-based clustering method that uses a t-distributed error model to identify spatial clusters that are more robust to the presence of outliers caused by technical noise.
BayesSpace can spatially resolve expression patterns to near single-cell resolution without the need for external single-cell sequencing data.
BayesSpace is a fully Bayesian spatial clustering method that models a low-dimensional representation of the gene expression matrix and encourages neighboring spots to belong to the same cluster via a spatial prior.

□ DTFLOW: Inference and Visualization of Single-cell Pseudo-temporal Trajectories Using Diffusion Propagation
>> https://www.biorxiv.org/content/10.1101/2020.09.10.290973v1.full.pdf
DTFLOW uses an innovative approach named Reverse Searching on kNN Graph (RSKG) to identify the underlying multi-branching processes of cellular differentiation.
DTFLOW infers the pseudo-time trajectories using single-cell data. DTFLOW uses a new manifold learning method, Bhattacharyya kernel feature decomposition (BKFD), for the visualization of underlying dataset structure.

□ Polee: RNA-Seq analysis using approximate likelihood
>> https://www.biorxiv.org/content/10.1101/2020.09.09.290411v1.full.pdf
Estimating transcript expression necessitates either ignoring ambiguous reads, explicitly assigning them to transcripts, or otherwise implicitly considering the space of possible assignments.
Pólya tree transformation, a new method of approximating the likelihood function of a sparse mixture model. a general approach to reducing the the computational demands of probabilistic RNA-Seq models, is a significant push in the direction of honest accounting of uncertainty.

□ OrbNet: Deep Learning for Quantum Chemistry Using Symmetry-Adapted Atomic-Orbital Features
>> https://arxiv.org/pdf/2007.08026.pdf
OrbNet, a machine learning method in which energy solutions from the Schrodinger equation are predicted using symmetry adapted atomic orbitals features and a graph neural-network architecture.
OrbNet outperforms existing methods in terms of learning efficiency and transferability for the prediction of density functional theory results while employing low-cost features that are obtained from semi-empirical electronic structure calculations.
The key elements of OrbNet incl. the efficient evaluation of the features in the symmetry-adapted atomic orbitals basis, the utilization of a graph-NN w/ edge and node attention and message passing layers, and a prediction phase that ensures extensivity of the resulting energies.

□ sem1R: Finding semantic patterns in omics data using concept rule learning with an ontology-based refinement operator
>> https://biodatamining.biomedcentral.com/articles/10.1186/s13040-020-00219-6
sem1R allows for the induction of more complex patterns of 2-dimensional binary omics data. This extension allows to discover and describe semantically coherent biclusters.
sem1R reveals interpretable hidden rules in omics data. These rules capture semantic differences b/w a target & a non-target class. the refinement operator uses Redundant Generalization & Redundant Non-potential, both of which dramatically prune the rule space and consequently.
□ Classification in biological networks with hypergraphlet kernels
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa768/5901538
a hypergraph-based approach for modeling biological systems and formulate vertex classification, edge classification and link prediction problems on (hyper)graphs as instances of vertex classification on (extended, dual) hypergraphs.
a novel kernel method on vertex- and edge-labeled (colored) hypergraphs for analysis and learning. The method is based on exact and inexact (via hypergraph edit distances) enumeration of hypergraphlets.

□ HyperXPair: Learning distance-dependent motif interactions: an interpretable CNN model of genomic events
>> https://www.biorxiv.org/content/10.1101/2020.08.27.270967v1.full.pdf
HyperXPair (the Hyper-parameter eXplainable Motif Pair framework), a new architecture that learns biological motifs and their distance-dependent context through explicitly interpretable parameters.
In the inner-loop, HyperXPair uses stochastic gradient descent to search for the motifs assumed distance-dependent interactions. In the outer-loop, HyperXpair uses Bayesian Optimization for the hyper-parameters {M,Σ}, where the network weights are learned anew at each iteration.
□ Single-cell mapper (scMappR): using scRNA-seq to infer cell-type specificities of differentially expressed genes
>> https://www.biorxiv.org/content/10.1101/2020.08.24.265298v1.full.pdf
single cell Mapper (scMappR), a method that assigns cell-type specificity scores to DEGs obtained from bulk RNA-seq by integrating cell-type expression data generated by scRNA-seq and existing deconvolution methods.
scMappR ensures that the cell-type specific expression is relevant to the inputted gene list by containing a bioinformatic pipeline to process scRNA-seq data into a signature matrix, and pre-computed signature matrices of reprocessed scRNA-seq data.
□ SPEARS: Standard Performance Evaluation of Ancestral Haplotype Reconstruction through Simulation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa749/5896985
SPEARS allows start-to-finish analysis of a given population through simulation with SAEGUS, imputation with MaCH, and ancestral haplotype reconstruction with RABBIT.
SPEARS determines the reliablability of inferred ancestral haplotype maps. This truth data is retained but also modified to mimic sparse genotype data that enters the standard multi-step process of imputation and haplotype reconstruction.

□ BSCET: Detecting cell-type-specific allelic expression imbalance by integrative analysis of bulk and single-cell RNA sequencing data
>> https://www.biorxiv.org/content/10.1101/2020.08.26.267815v1.full.pdf
BSCET, which enables the characterization of cell-type-specific AEI in bulk RNA-seq data by integrating cell type composition information inferred from a small set of scRNA-seq samples, possibly obtained from an external dataset.
the allelic read counts were modeled using a generalized linear model (GLM) for binomial data with logit link function.
The relative expression of the reference allele over total read counts was modeled across individuals using an intercept only model, and evidence of AEI was assessed by testing whether the intercept is significantly different from zero.
□ singleCellHaystack: A clustering-independent method for finding differentially expressed genes in single-cell transcriptome data
>> https://www.nature.com/articles/s41467-020-17900-3
singleCellHaystack does not rely on clustering, thus, avoiding biases caused by the arbitrary clustering of cells. singleCellHaystack found large numbers of statistically significant DEGs in all datasets.
singleCellHaystack uses Kullback–Leibler divergence to find genes that are expressed in subsets of cells that are non-randomly positioned in a multidimensional space.
□ GLPCA: Constrained Clustering With Dissimilarity Propagation-Guided Graph-Laplacian PCA
>> https://ieeexplore.ieee.org/document/9178787
the dissimilarity propagation-guided graph-Laplacian principal component analysis (DP-GLPCA) is capable of capturing both the local and global structures of input samples to exploit their characteristics for excellent clustering.
a convex semisupervised low-dimensional embedding model by incorporating a new dissimilarity regularizer into GLPCA, in which both the similarity and dissimilarity between low-dimensional representations are enforced with the constraints to improve their discriminability.
□ Hyperbolic geometry of gene expression
>>
a non-metric multidimensional embedding (MDS) in hyperbolic space which, combined with Euclidean MDS, can quantitatively detect intrinsic geometry and characterize its properties.
When using the metric MDS, the Shepard diagram shows increased spread but does not yield a nonlinear relationship when embedding Euclidean data to hyperbolic space.
The reason is that Euclidean distances can be embedded into the faster-expanding hyperbolic space masking the distortion of distances, and this does not happen in the non-metric MDS.
□ MixGenotype: A machine learning framework for genotyping the structural variations with copy number variant
>> https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-020-00733-w
The Multiclass Relevance Vector Machine (M-RVM) was combined with the distribution characteristics of the features. M-RVM can efficiently deal with the problem of low-dimensional linear inseparability and output the result of genotyping with the greatest possibility.
□ CSS: cluster similarity spectrum integration of single-cell genomics data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02147-4
CSS can be used to assess cellular heterogeneity and enable reconstruction of differentiation trajectories from cerebral organoid and other single-cell transcriptomic data.
CSS considers every cell cluster in each sample as an intrinsic reference for integration and represents each cell by its transcriptome’s similarity to clusters across samples. CSS allows query data projection to the reference scRNA-seq atlas.

□ GraphSCC: Accurately Clustering Single-cell RNA-seq data by Capturing Structural Relations between Cells through Graph Convolutional Network
>>
a denoising autoencoder network was employed to obtain low dimensional representations for capturing local structural, and a dual self-supervised module was utilized to optimize the representations and the clustering objective function iteratively in an unsupervised manner.
GraphSCC is able to effec- tively capture the relations between cells and the characteristics of data by learning representations using the GCN and DAE modules. GraphSCC provides representations for better intra-cluster compactness and inter-cluster separability.

□ SCENA: Correlation imputation in single cell RNA-seq using auxiliary information and ensemble learning
>> https://www.biorxiv.org/content/10.1101/2020.09.03.282178v1.full.pdf
The SCENA gene-by-gene correlation matrix estimate is obtained by model stacking of multiple imputed correlation matrices based on known auxiliary information about gene connections.
SCENA not only accurately imputes gene correlations but also outperforms existing imputation approaches in downstream analyses such as dimension reduction, cell clustering, graphical model estimation.
□ starmapVR: immersive visualisation of single cell spatial omic data
>> https://www.biorxiv.org/content/10.1101/2020.09.01.277079v1.full.pdf
starmapVR enables single cell multivariate data to be visualised alongside the spatial cues, such as a histological image that match the layout of the cells, in a two and a half dimensional (2.5D) visualisation strategy.
starmapVR can support visualise exploration of large single cell data in their inferred or actual spatial context.
□ EXFI: Exon and splice graph prediction without a reference genome
>> https://onlinelibrary.wiley.com/doi/full/10.1002/ece3.6587
Get exons from a transcriptome and raw genomic reads using abyss-bloom and bedtools. EXFI predicts the splice graph and exon sequences using an assembled transcriptome and raw whole‐genome sequencing reads.
The main algorithm uses Bloom filters to remove reads that are not part of the transcriptome, to predict the intron–exon boundaries, to then proceed to call exons from the assembly, and to generate the underlying splice graph.
the remaining reads are used to build a cascading Bloom filter with ABySS. The results are returned in GFA1 format, which encodes both the predicted exon sequences and how they are connected to form transcripts.
□ CPE-SLDI: Predicting Coding Potential of RNA Sequences by Solving Local Data Imbalance
>> https://ieeexplore.ieee.org/document/9186774
analyzing the distribution of ORF length of RNA sequences, and observe that the number of coding RNAs with sORF is inadequate and coding RNAs with sORF are much less than ncRNAs with sORF.
CPE-SLDI constructs a prediction model by combining various sequence-derived features based on the augmented data.

□ ICICLE-seq: Integrated single cell analysis of chromatin accessibility and cell surface markers
>> https://www.biorxiv.org/content/10.1101/2020.09.04.283887v1.full.pdf
ICICLE-seq, Integrated Cellular Indexing of Chromatin Landscape and Epitopes combines cell surface marker barcoding with high quality scATAC-seq offers a novel tool to identify type-specific regulatory regions based on phenotypically defined cell types.

□ STRONG: Metagenomics Strain Resolution on Assembly Graphs
>> https://www.biorxiv.org/content/10.1101/2020.09.06.284828v1.full.pdf
STrain Resolution ON assembly Graphs (STRONG) avoids the limitations of the variant-based approaches by resolving haplotypes directly on assembly graphs using a novel variational Bayesian algorithm, BayesPaths.
BayesPaths allows more complex variant structure and read information. The BayesPaths is also a substantial algorithmic advance enabling coverage across multiple samples to be incorporated into a rigorous Bayesian procedure that gives uncertainties in both the strain abundances.
□ TERL: classification of transposable elements by convolutional neural networks
>> https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbaa185/5900933
TERL - Transposable Elements Representation Learner preprocesses and transforms one-dimensional sequences into two-dimensional space data.
TERL can learn how to predict any hierarchical level of the TEs classification system and is about 20 times and three orders of magnitude faster than TEclass and PASTEC.
□ SAIGEgds – an efficient statistical tool for large-scale PheWAS with mixed models
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa731/5902828
The package implements the SAIGE method in optimized C ++ codes, taking advantage of sparse genotype dosages and integrating the efficient genomic data structure (GDS) file format.
SAIGEgds is designed for single variant tests in large-scale phenome-wide association studies (PheWAS) with millions of variants and samples, controlling for sample structure and case-control imbalance.

□ BURST enables mathematically optimal short-read alignment for big data
>> https://www.biorxiv.org/content/10.1101/2020.09.08.287128v1.full.pdf
BURST, a high-throughput DNA short-read aligner that uses several new synergistic optimizations to enable provably optimal alignment.
Although BURST is up to 20,000 times faster than previous optimal gapped alignment algorithms, it will still be 10-to-100 times slower than some heuristic or non-optimal search methods such as k-mer-based search.

□ phASER: A vast resource of allelic expression data spanning human tissues
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02122-z
the utility of a vast AE resource generated from the GTEx v8 release, containing 15,253 samples spanning 54 human tissues for a total of 431 million measurements of AE at the SNP level and 153 million measurements at the haplotype level.
an extension of phASER that allows effect sizes of cis-regulatory variants to be estimated using haplotype-level AE data. as a result of improvements in genome phasing, data can be aggregated across SNPs to produce estimates of AE at the haplotype level.
phASER does this systematically, in a way that uses the information contained within reads to improve phasing, while preventing double counting of reads across SNPs to improve the quality of data generated.
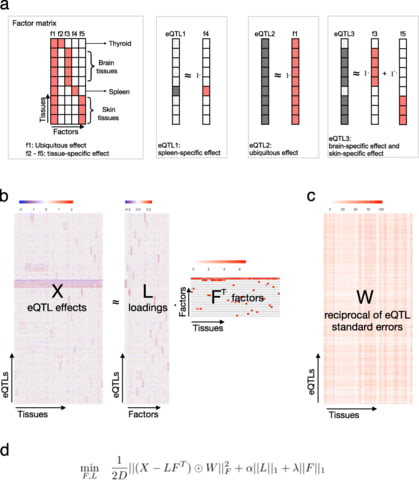
□ sn-spMF: matrix factorization informs tissue-specific genetic regulation of gene expression
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02129-6
sn-spMF, a constrained matrix factorization model called weighted semi-nonnegative sparse matrix factorization and apply it to analyze eQTLs across 49 human tissues from the GTEx consortium.
sn-spMF identifies larger numbers of ts-eQTLs that remain biologically coherent, an opportunity for novel mechanistic insights. Different applications, such as time series or perturbation-response eQTL, may ultimately benefit from specialized matrix factorization formulations.

□ Rapid Development of Improved Data-dependent Acquisition Strategies
>> https://www.biorxiv.org/content/10.1101/2020.09.11.293092v1.full.pdf
a framework to support this development process by extending the capability of ViMMS so it could easily run fragmentation strategies implemented as controllers in the simulator on the real MS equipment with minimal change to the code.
WeightedDEW generalises the dynamic exclusion window approach to a real-valued weighting scheme allowing previously fragmented ions to smoothly rise up the priority list as their intensity remains high.

□ nanoDoc: RNA modification detection using Nanopore raw reads with Deep One-Class Classification
>> https://www.biorxiv.org/content/10.1101/2020.09.13.295089v1.full.pdf
nanoDoc detects PTMs from from non-modified in vitro signaling could be solved by One-Class Classification. Current signal deviations caused by PTMs are analyzed via Deep One-Class Classification with a convolutional neural network.
The 16-dimensional feature output of the Native sequence was subjected to k-mean clustering and dimensional reduction uniform manifold approximation and projection.