








 (“Knight in the Dark | Envy Avenue”)
(“Knight in the Dark | Envy Avenue”)『無知は武器となる』 ー 観測し得る情報の欠損こそが、完全性を指向する意思のベクトルを決定し、
起こるべくして起こることを知らないということ自体が、揺るぎない主義と人間性を規定する。

□ ReDX: Repeated Decision Stumping Distils Simple Rules from Single Cell Data
>> https://www.biorxiv.org/content/10.1101/2020.09.08.288662v1.full.pdf
Repeated Decision Stumping (ReDX) that exploits the simple structure of decision tree models to distill highly interpretable but still predictive insights from single cell data single genes or sets of genes that show particular statistical association with developmental events.
ReDX sits between these two types of algorithms and allows us to generate new interpretable hypotheses and mechanistic models in a data-driven framework.
Whilst each learnt model is simple and 1-dimensional, ReDX demonstrates their ability to segregate cells along diverse developmental boundaries with great precision. Further, as a pragmatic method for gaining insights, maybe even intuition, about a high-dimensional system.

□ OGRE: Overlap Graph-based metagenomic Read clustEring
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa760/5900259
OGRE outperforms other read binners in terms of the number of species included in a cluster, also referred to as cluster purity, and the fraction of all reads that is placed in one of the clusters.
OGRE constructs an overlap graph using Minimap2, filters out a large fraction of the overlaps b/w reads from different species, clusters reads using single linkage clustering, and merges highly similar clusters. Even though these processes all have low computational complexity.

□ scGCN: a Graph Convolutional Networks Algorithm for Knowledge Transfer in Single Cell Omics
>>
scGCN learns a sparse and hybrid graph of both inter- and intra-dataset cell mappings using mutual nearest neighbors of canonical correlation vectors. scGCN projects different datasets onto a correlated low-dimensional space.
scGCN nonlinearly propagates feature information from neighboring cells in the hybrid graph, which learns the topological cell relations and improves the performance of transferring labels by considering higher-order relations between cells.

□ LIQA: Long-read Isoform Quantification and Analysis
>> https://www.biorxiv.org/content/10.1101/2020.09.09.289793v1.full.pdf
LIQA incorporates base-pair quality score and isoform-specific read length information to assign different weights across reads, which reflects alignment confidence.
LIQA is computationally intensive because the approximation of non-parametric Kaplan-Meier estimator of function relies on empirical read length distribution and the parameters are estimated using EM-algorithm.

□ Watershed: Transcriptomic signatures across human tissues identify functional rare genetic variation
>> https://science.sciencemag.org/content/369/6509/eaaz5900.full
Using 838 samples with whole-genome and multitissue transcriptome sequencing data in the Genotype-Tissue Expression (GTEx) project version 8, they assessed how rare genetic variants contribute to extreme patterns in gene expression, allelic expression, and alternative splicing.
Watershed, a probabilistic model for personal genome interpretation that improves over standard genomic annotation–based methods for scoring RVs by integrating these three transcriptomic signals from the same individual and replicates in an independent cohort.
Watershed automatically learns Markov random field (MRF) edge weights reflecting the strength of the relationship between the different tissues or phenotypes included that together allow the model to predict functional effects accurately.
□ VALERIE: Visual-based inspection of alternative splicing events at single-cell resolution
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008195
VALERIE generates an ensemble of informative plots to visualise cell-to-cell heterogeneity of alternative splicing profiles across single cells and performs statistical tests to compare percent spliced-in (PSI) values across the user-defined groups of cells.
VALERIE complements existing implementations by enabling visualisation of ASE for scRNA-seq data typically generated by full-length library preparation methods such as Smart-seq2 and it is not appropriate for high-throughput droplet-based platforms such as the Chromium 10x.

□ CABEAN: A Software for the Control of Asynchronous Boolean Networks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa752/5897411
CABEAN integrates several methods for the source-target control of asynchronous Boolean networks.
CABEAN utilises MCMAS to encode Boolean networks into the efficient data structure binary decision diagram (BDD). CABEAN identifies efficacious nodes, whose perturbations can drive the dynamics of a network from a source attractor to a target attractor.

□ Effects of underlying gene-regulation network structure on prediction accuracy in high-dimensional regression
>> https://www.biorxiv.org/content/10.1101/2020.09.11.293456v1.full.pdf
When a limited number of regression coefficients had a large contribution to the definition of a trait, and the gene regulation network was random (Σ2), the simulation indicated that models with high estimation accuracy could be developed from a small number of observations.
a real gene regulation network is likely to exhibit scale-free structure. As the lasso-type regularization methods shrink parameters toward zero, the correlations among exploratory variables reduce with the graphical lasso.
□ MR-Clust: Clustering of genetic variants in Mendelian randomization with similar causal estimates
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa778/5904264
While the hypothesis of whether a risk factor has a causal effect on an outcome can be assessed with a single valid IV, most genetic variants do not explain enough variability in the risk factor to have sufficient power to reliably detect a moderate-sized causal effect.
MR-Clust, a method in the context of Mendelian randomization that clusters genetic variants associated with a given risk factor according to the variant’s associations with the risk factor and outcome. MR-Clust identifies variants that reflect distinct causal mechanisms.
□ FH-Clust: Data integration by fuzzy similarity-based hierarchical clustering
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03567-6
The idea behind the proposed approach comes from observing that a hierarchical clustering dendrogram can be associated with a Łukasiewicz fuzzy similarity-based equivalence relation.
So that a consensus matrix, that is the representative information of all dendrograms, is derived by combining multiple hierarchical agglomerations following an approach based on transitive consensus matrix construction.

□ RabbitMash: Accelerating hash-based genome analysis on modern multi-core architectures
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa754/5897409
RabbitMash is an efficient highly optimized implementation of Mash which can take full advantage of modern hardware including multi-threading, vectorization, and fast I/O.
RabbitMash is able to compute the all-vs-all distances of 100,321 genomes in less than 5 minutes on a 40-core workstation while Mash requires over 40 minutes.

□ Genome-wide identification of genes regulating DNA methylation using genetic anchors for causal inference
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02114-z
The genome-wide analysis, utilizing genetic instruments for gene expression, identified 818 genes that affect distant DNA methylation levels in blood and provide insights into the principles of epigenetic regulation.
By employing genetic instruments as causal anchors, the directed associations between gene expression and distant DNA methylation levels, while ensuring specificity of the associations by correcting for linkage disequilibrium and pleiotropy among neighboring genes.

□ MCMSeq: Bayesian hierarchical modeling of clustered and repeated measures RNA sequencing experiments
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03715-y
MCMSeq best combines high statistical power (i.e. sensitivity or recall) with maintenance of nominal false positive and false discovery rates compared the other available strategies, especially at the smaller sample sizes investigated.
MCMseq fits the NBGLMM as a Bayesian hierarchical model using MCMC. Both the NBGLMM and CPGLMM consistently had highly inflated type 1 error rates and FDRs when fit under a frequentist paradigm.

□ GO2PLS: Statistical Integration of Multiple Omics Datasets
>>
Two-way Orthogonal Partial Least Squares (O2PLS) captures the heterogeneity by introducing the orthogonal subspaces and better estimates the joint subspaces.
GO2PLS-based methods generally outperformed PCA and PLS-based methods regarding joint score estimation when orthogonal variation. GO2PLS is efficient in estimating latent components that represent underlying systems.

□ scMontage: Fast and Robust Gene Expression Similarity Search for Massive Single-cell Data
>> https://www.biorxiv.org/content/10.1101/2020.08.30.271395v1.full.pdf
The scMontage also provides quick access to additional information of various cell types in the SHOGoiN database from the search results.
The scMontage search is based on Spearman's rank correlation coefficient as a similarity metric of gene expression profiles using a very fast algorithm, RaPiDS, and its robustness is ensured by introducing Fisher's Z-transformation and Z-test.
□ Graphia: A platform for the graph-based visualisation and analysis of complex data
>> https://www.biorxiv.org/content/10.1101/2020.09.02.279349v1.full.pdf
Where a graph has been generated from a numerical matrix, it will also automatically calculate the maximum, minimum mean and variance of the data series represented by a node.
Graphia incorporates the k-NN algorithm, which culls all but the top k edges, according to the value of a nominated attribute. Graphia also incorporates the MCL and Louvain clustering algorithms, where the granularity of clustering can be adjusted after their initial calculation.

□ PLAST: Detecting High Scoring Local Alignments in Pangenome Graphs
>> https://www.biorxiv.org/content/10.1101/2020.09.03.280958v1.full.pdf
PLAST, a new heuristic method to find maximum scoring local alignments of a DNA query sequence to a pangenome represented as a compacted colored de Bruijn graph.
similarly to the statistical behavior of pairwise alignment, sequence-to-graph alignment p-values exhibit exponential tails, log ps ≈ C − λ · s for constants C ∈ R, λ > 0.

□ EPISCORE: cell type deconvolution of bulk tissue DNA methylomes from single-cell RNA-Seq data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02126-9
EPISCORE, a computational algorithm that performs virtual microdissection of bulk tissue DNA methylation data at single cell-type resolution for any solid tissue.
EPISCORE applies a probabilistic epigenetic model of gene regulation to a single-cell RNA-seq tissue atlas to generate a tissue-specific DNA methylation reference matrix.
□ TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads
>> https://academic.oup.com/gigascience/article/9/9/giaa094/5902284
TGS-Gapcloser enables the combination of different genetic information with different lengths and resolutions and makes it possible to complete high-quality (ultra) large genome assemblies.
Using only 10× coverage of ONT or PacBio long reads applied to 3 de novo assembled human genomes, TGS-GapCloser increases in the scaftig NG50 by 11.0- to 45.0-fold and an increase in the scaftig NGA50 by 6.8- to 30.6-fold.
□ Linguae Naturalis Principia Mathematica: A mathematical model for universal semantics
>> https://ieeexplore.ieee.org/document/9187687
The Markov semantic model allows us to represent each topical concept by a low-dimensional vector, interpretable as algebraic invariants in succinct statistical operations on the document, targeting local environments of individual words.
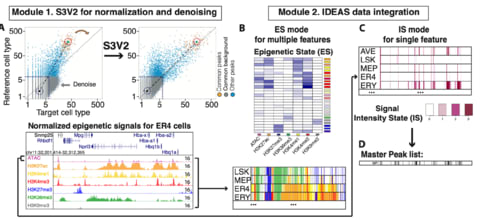
□ S3V2-IDEAS: a package for normalizing, denoising and integrating epigenomic datasets across different cell types
>> https://www.biorxiv.org/content/10.1101/2020.09.08.287920v1.full.pdf
S3V2-IDEAS identifies epigenetic states for multiple features, or identify signal intensity states and a master peak list across different cell types for a single feature.
The S3V2-IDEAS produces three outputs: the normalized signal tracks and the -log10 p-value tracks based on the background model; a list of epigenetic states or signal intensity states and the corresponding state track in each cell type.
□ Target controllability with minimal mediators in complex biological networks
>> https://www.sciencedirect.com/science/article/pii/S0888754320311861
the path length is a major determinant of in properties of the target control under minimal mediators. As the average path length becomes larger, the ratio of drivers to target nodes decreases and the ratio of mediators to targets increases.
Target Controllability with Minimal Mediators in Complex Biological Networks. The proposed methodology has potential applications in any directed networks, Based on path lengths between node pairs and meets Kalman’s controllability rank condition.
□ LongGeneDB: a data hub for long genes
>> https://www.biorxiv.org/content/10.1101/2020.09.08.281220v1.full.pdf
Long genes harbor specific genomic and epigenomic features and have been implicated in many humaんdiseases. LongGeneDB is an interactive, visual database containing genomic information of 992 long genes (>200 kb) in 15 species.
LongGeneDB normalizes each profile by their sequencing depth to obtain the reads per million uniquely mapped reads values, merged the biological replicates, calculated the merged profiles of the gene region and the upstream and downstream 100kb regions of each long gene.
□ Incorporating prior knowledge into regularized regression
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa776/5904263
the proposed regression with individualized penalties can outperform the standard LASSO in terms of both parameters estimation and prediction performance when the external data is informative.
a new penalized regression approach that allows a-priori integration of external meta-features. The method extends LASSO regression by incorporating individualized penalty parameters for each regression coefficient.
□ PTWAS: investigating tissue-relevant causal molecular mechanisms of complex traits using probabilistic TWAS analysis
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02026-y
probabilistic TWAS (PTWAS) provides novel functionalities to evaluate the causal assumptions and estimate tissue- or cell-type specific causal effects of gene expression on complex traits.
PTWAS is built upon the causal inference framework of IV analysis, and utilizes probabilistic eQTL annotations derived from multi-variant Bayesian fine-mapping analysis conferring higher power to detect TWAS associations than existing methods.

□ An extended catalogue of tandem alternative splice sites in human tissue transcriptomes
>> https://www.biorxiv.org/content/10.1101/2020.09.11.292722v1.full.pdf
The significantly expressed miSS evolve under the same selection pressure as maSS, while other miSS lack signatures of evolutionary selection and conservation.
a zero-inflated Poisson linear model that describes the dependence of miSS-specific read counts (rmin) on maSS-specific read counts (rmaj).
□ Diversification of reprogramming trajectories revealed by parallel single-cell transcriptome and chromatin accessibility sequencing
>> https://advances.sciencemag.org/content/6/37/eaba1190/tab-figures-data
Binary choice between a FOSL1 and a TEAD4-centric regulatory network determines the outcome of a successful reprogramming.
the single-cell roadmap of the human cellular reprogramming process, which reveals the diverse cell fate trajectory of individual reprogramming cells.

□ Shark: fishing relevant reads in an RNA-Seq sample
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaa779/5905477
Shark, a fast tool for mapping-free gene separation of reads, using Bloom filter. Shark relies on succinct data structures and multi-threading.
Shark provides a preprocessing step that significantly reduces the running time and/or the memory requirements of computationally-intensive downstream analyses, while not negatively impacting their results. Shark is the specifically designed for computing a gene assignment.

□ phasedibd: Fast and robust identity-by-descent inference with the templated positional Burrows-Wheeler transform
>> https://www.biorxiv.org/content/10.1101/2020.09.14.296939v1.full.pdf
phasedibd computes phase aware identity-by-descent (IBD) using the templated positional Burrows-Wheeler transform (TPBWT). The TPBWT is an extension of the PBWT with an extra dimension added that masks out potential errors in the haplotypes and extends IBD segments through putative errors.
Any TPBWT-based algorithms for phasing and/or imputation could be designed to run directly over TPBWT-compressed haplotypes making large scale reference-based estimates computationally tractable.

□ Primo: integration of multiple GWAS and omics QTL summary statistics for elucidation of molecular mechanisms of trait-associated SNPs and detection of pleiotropy in complex traits
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02125-w
Primo allows arbitrary study heterogeneity and detects coordinated effects from multiple studies while not requiring the effect sizes to be the same, it allows the summary statistics to be calculated with independent or overlapping samples with unknown sample correlations.
Primo examines the conditional associations of a known trait-associated SNP with other complex and omics traits adjusting for other lead SNPs in a gene region.
Primo moves beyond joint association towards colocalization and provides a thorough inspection of the effects of multiple SNPs within a region to reduce spurious associations due to linkage disequilibrium.

□ ELRF: A Linear-Time Solution to the Labeled Robinson-Foulds Distance Problem
>> https://www.biorxiv.org/content/10.1101/2020.09.14.293522v1.full.pdf
a different formulation of the Labeled Robinson Foulds (LRF) edit distance — based on node insertion, deletion and label substitution — comparing two node-labeled trees, which can be computed in linear time.
The new formulation maintains other desirable properties: being a metric, reducing to RF for unlabeled trees and maintaining an intuitive interpretation. The LRF distance overcomes the major drawback of ELRF , namely the lack of an exact polynomial algorithm for the latter.
the ability of LRF to support an arbitrary number of labels makes it applicable to gene trees containing more than just speciations and duplications, such as horizontal gene transfers or gene conversion events.

□ HiSCF: leveraging higher-order structures for clustering analysis in biological networks
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa775/5906025
HiSCF, a novel clustering framework to identify functional modules based on the higher-order structure information available in a biological network.
Taking advantage of higher-order Markov stochastic process, HiSCF is able to perform the clustering analysis by exploiting a variety of network motifs.
□ SEEDS: Data driven inference of structural model errors and unknown inputs for dynamic systems biology
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btaa786/5906024
Inaccurate knowledge about the structure, functional form and parameters of interactions is a major obstacle to mechanistic modelling.
SEEDS estimate Hidden Inputs using the Dynamic Elastic Net. Algorithms to calculate the hidden inputs of systems of differential equations.
□ Seagull: lasso, group lasso and sparse-group lasso regularization for linear regression models via proximal gradient descent
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03725-w
To simultaneously detect non-zero effects and account for the relatedness of explanatory variables, the lasso has been modified and enhanced to the group lasso, the sparse-group lasso and the “Integrative LASSO with Penalty Factors” (IPF-lasso).
Seagull, a fast and numerically cheap implementation of these operators via proximal gradient descent. Seagull computed the solution in a fraction of the time needed by SGL.
□ ADEPT: a domain independent sequence alignment strategy for gpu architectures
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03720-1
ADEPT has a novel data structure to tackle the inter-thread dependencies and utilized register-to-register data transfers for efficient communication between CUDA threads.
ADEPT, a novel domain independent sequence alignment strategy for GPU architectures for accelerating dynamic programming based sequence alignment algorithms. ADEPT shows that a GPU-accelerated complete Smith-Waterman algorithm for the use case of pairwise sequence alignments.

□ TIMEOR: a web-based tool to uncover temporal regulatory mechanisms from multi-omics data
>> https://www.biorxiv.org/content/10.1101/2020.09.14.296418v1.full.pdf
TIMEOR (Trajectory Inference and Mechanism Exploration with Omics data in R) addresses the critical need for methods to predict causal regulatory mechanism networks between TFs from time series multi-omics data.
TIMEOR merges experimentally determined gene networks, time series RNA- seq and motif and ChIP-seq information to reconstruct TF GRNs with directed causal interaction edges by labeling the causal interaction and regulation between gene regulatory events across time.

□ CIAlign - A highly customisable command line tool to clean, interpret and visualise multiple sequence alignments
>> https://www.biorxiv.org/content/10.1101/2020.09.14.291484v1.full.pdf
CIAlign effectively removes poorly aligned regions and sequences from MSAs and provides novel visualisation options. The tool is aimed at anyone who wishes to automatically clean up parts of an MSA and those requiring a new, accessible way for visualising large MSAs.
When running CIAlign with all core functions and for fixed gap proportions, the runtime scales quadratically with the size of the MSA, i.e. with n as the number of sequences and m the length of the MSA, the worst case time complexity is O((nm)2).