「種 (species)」とは、力学的平衡状態にある均質個体群のトポロジーな偏りを持つ複製発生確率の連続体であり、あるいはそのような位相同型からなる物性の時間保存性を有する概念上の分類である。
自然や宇宙、生命の悠久の営みは、時として大きな犠牲を無作為に、意にも介せず一いとも簡単に奪い去ってしまう。
この途方もない渦流の中で、名も無き私たちが「誰か」であることはまるで無意味に思える。
しかし、時は精細な構造物であり、私たちが誰かであり、何を為すのかは複雑な力学的共時性に在る。
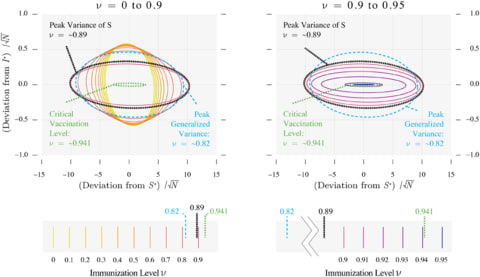
□ The statistics of epidemic transitions
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006917
“rooted in dynamical systems and the theory of stochastic processes” have yielded insight into the dynamics of emerging and re-emerging pathogens.
This perspective views pathogen emergence and re-emergence as a “critical transition,” and uses the concept of noisy dynamic bifurcation to understand the relationship between the system observables and the distance to this transition.

□ Morphoseq: – a shorter way to longer reads
>> http://longastech.com
Morphoseq is a "virtual long read" library preparation technology that computationally increases read length.
Morphoseq is a disruptive technology to convert short read sequencers into ‘virtual long read’ sequencers, enabling finished quality genome assemblies with high accuracy, including resolution of difficult-to-assemble genomic regions. Morphoseq utilises a proprietary mutagenesis reaction to introduce unique mutation patterns into long DNA molecules, up to 10 kbp and greater.
The custom Morphoseq algorithm uses the unique identifiers to reconstruct the original long DNA template sequences. The resulting long DNA sequences are extremely high quality, with typical accuracy in excess of 99.9%.
□ Morphoseq: Longas Technologies Launches, Offering 'Virtual Long Read' Library Prep
>> https://www.genomeweb.com/sequencing/longas-technologies-launches-offering-virtual-long-read-library-prep
Morphoseq Novel DNA Sequencing Technology Enables High Accurate and Cost Effective Long Read Sequencing on Short Read NGS Platforms
>> http://longastech.com/morphoseq-novel-dna-sequencing-technology-enables-highly-accurate-and-cost-effective-long-read-sequencing-on-short-read-ngs-platforms/
Aaron Darling demonstrated long reads up to 15kbp with modal accuracy 100% and 92% of reads >Q40 when measured against independent reference genomes.
These results, on a set of 60 multiplexed bacterial isolates show that genomic coverage is highly uniform with the data yielding finished-quality closed circle assemblies for bacterial genomes across the entire GC content range.
Morphoseq effectively converts short read sequencers into virtual ‘long read’ sequencers, enabling finished-quality genome assemblies with high accuracy, including resolution of difficult-to-assemble genomic regions.
□ DarkDiv: Estimating probabilistic dark diversity based on the hypergeometric distribution
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/15/636753.full.pdf
DarkDiv is a novel method based on the hypergeometric probability distribution to assign probabilistic estimates of dark diversity.
Future integration of probabilistic species pools and functional diversity will advance our understanding of assembly processes and conservation status of ecological systems at multiple spatial and temporal scales.

□ Pairwise and higher-order genetic interactions during the evolution of a tRNA
>> https://www.nature.com/articles/s41586-018-0170-7
Notably, all pairs of mutations interacted in at least 9% of genetic backgrounds and all pairs switched from interacting positively to interacting negatively in different genotypes.
Higher-order interactions are also abundant and dynamic across genotypes. The epistasis in this tRNA means that all individual mutations switch from detrimental to beneficial, even in closely related genotypes.
As a consequence, accurate genetic prediction requires mutation effects to be measured across different genetic backgrounds and the use of higher-order epistatic terms.

□ SISUA: SemI-SUpervised generative Autoencoder for single cell data:
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/08/631382.full.pdf
assuming the true data manifold is of much lower-dimension than the embedded dimensionality of the data. Embedded-dimensionality De in this case is the number of selected genes in a single scRNA-seq vector xi of a cell i.
SISUA model based on the Bayesian generative approach, where protein quantification available as CITE-seq counts from the same cells are used to constrain the learning process. The generative model is based on the deep variational autoencoder (VAE) neural network architecture.

□ Proteome-by-phenome Mendelian Randomisation detects 38 proteins with causal roles in human diseases and traits
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/10/631747.full.pdf
confirmatory evidence for a causal role for the proteins encoded at multiple cardiovascular disease risk loci (FGF5, IL6R, LPL, LTA), and discovered that intestinal fatty acid binding protein (FABP2) contributes to disease pathogenesis.
applying pQTL based MR in a data-driven manner across the full range of phenotypes available in GeneAtlas, as well as supplementing this with additional studies identified through Phenoscanner.

□ SCRABBLE: single-cell RNA-seq imputation constrained by bulk RNA-seq data:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1681-8
Single-cell RNA-seq data contain a large proportion of zeros for expressed genes. Such dropout events present a fundamental challenge for various types of data analyses.
SCRABBLE leverages bulk data as a constraint and reduces unwanted bias towards expressed genes during imputation. SCRABBLE outperforms the existing methods in recovering dropout events, capturing true distribution of gene expression across cells, and preserving gene-gene relationship and cell-cell relationship in the data.
SCRABBLE is based on the framework of matrix regularization that does not impose an assumption of specific statistical distributions for gene expression levels and dropout probabilities.

□ A New Model for Single-Molecule Tracking Analysis of Transcription Factor Dynamics
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/14/637355.full.pdf
an improved method to account for photobleaching effects, theory-based models to accurately describe transcription factor dynamics, and an unbiased model selection approach to determine the best predicting model.
The continuum of affinities model. TFs can diffuse on the DNA, and transition between any state (Diffusive, specifically bound, nonspecifically bound). Dwell time is defined as the time spent on the DNA, either bound or sliding.
A new interpretation of transcriptional regulation emerges from the proposed models wherein transcription factor searching and binding on the DNA results in a broad distribution of binding affinities and accounts for the power-law behavior of transcription factor residence times.

□ Artifacts in gene expression data cause problems for gene co-expression networks
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1700-9
for scale-free networks, principal components of a gene expression matrix can consistently identify components that reflect artifacts in the data rather than network relationships. Several studies have employed the assumption of scale-free topology to infer high-dimensional gene co-expression and splicing networks.
theoretically, in simulation, and empirically, that principal component correction of gene expression measurements prior to network inference can reduce false discoveries.

□ Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population
>> https://www.mdpi.com/2073-4425/10/5/352
Satellite sequence variation in the human genome is often so large that it is detected cytogenetically, yet due to the lack of a reference assembly and informatics tools to measure this variability,
contemporary high-resolution disease association studies are unable to detect causal variants in these regions. there is a pressing and unmet need to detect and incorporate this uncharacterized sequence variation into broad studies of human evolution and medical genomics.
□ Integration of Structured Biological Data Sources using Biological Expression Language
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/08/631812.full.pdf
BEL has begun to prove itself as a robust format in the curation and integration of previously isolated biological data sources of high granular information on genetic variation, epigenetics, chemogenomics, and clinical biomarkers.
Its syntax and semantics are also appropriate for representing, for example, disease-disease similarities, disease-protein associations, chemical space networks, genome-wide association studies, and phenome-wide association studies.
□ Regeneration Rosetta: An interactive web application to explore regeneration-associated gene expression and chromatin accessibility
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/08/632018.full.pdf
Regeneration Rosetta using either built-in or user-provided lists of genes in one of dozens of supported organisms, and facilitates the visualization of clustered temporal expression trends; identification of proximal and distal regions of accessible chromatin to expedite downstream motif analysis; and description of enriched functional gene ontology categories.
Regeneration Rosetta is broadly useful for both a deep investigation of time-dependent regulation during regeneration and hypothesis generation.
□ Random trees in the boundary of Outer space
>> https://arxiv.org/pdf/1904.10026v1.pdf
a complete understanding of these two properties for a “random” tree in ∂CVr. As a significant point of contrast to the surface case, and find that such a random tree of ∂CVr is not geometric.
the random walk induces a naturally associated hitting or exit measure ν on ∂CVr and that ν is the unique μ-stationary probability measure on ∂CVr, and ν gives full measure to the subspace of trees in ∂CVr which are free, arational, and uniquely ergodic.

□ The Energetics of Molecular Adaptation in Transcriptional Regulation
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/15/638270.full.pdf
a biophysical model of allosteric transcriptional regulation that directly links the location of a mutation within a repressor to the biophysical parameters that describe its behavior. explore the phenotypic space of a repressor with mutations in either the inducer binding or DNA binding domains.
Linking mutations to the parameters which govern the system allows for quantitative predictions of how the free energy of the system changes as a result, permitting coarse graining of high-dimensional data into a single-parameter description of the mutational consequences.
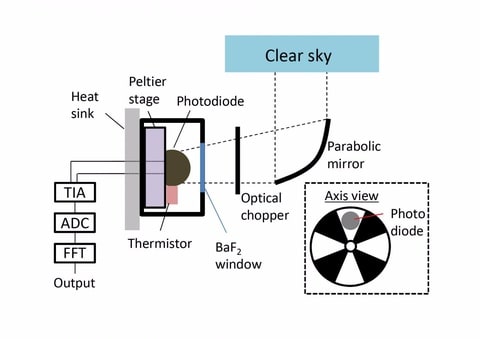
□ Experimental Device Generates Electricity from the Coldness of the Universe
>> https://www.ecnmag.com/news/2019/05/experimental-device-generates-electricity-coldness-universe
a device on Earth facing space, the chilling outflow of energy from the device can be harvested using the same kind of optoelectronic physics. “In terms of optoelectronic physics, there is really this very beautiful symmetry between harvesting incoming radiation and harvesting outgoing radiation.”
By pointing their device toward space, whose temperature approaches mere degrees from absolute zero.

□ p-bits for probabilistic spin logic
>> https://aip.scitation.org/doi/full/10.1063/1.5055860
The p-bit also provides a conceptual bridge between two active but disjoint fields of research, namely, stochastic machine learning and quantum computing.
First, there are the applications that are based on the similarity of a p-bit to the binary stochastic neuron (BSN), a well-known concept in machine learning.
□ evantthompson:
Friston's free-energy principle is based on the premise that living systems are ergodic. Kauffman begins his new book with the premise that life is non-ergodic. Who is right? My money is on Kauffman on this one, but what do I know? A World Beyond Physics https://global.oup.com/academic/product/a-world-beyond-physics-9780190871338
□ seanmcarroll:
Different things, no? Kauffman emphasizes that evolution of the genome is non-ergodic, which is certainly true. Friston only needs, presumably, the evolution of brain states to be ergodic. That's plausible, it's a much smaller space.
□ Degenerations of spherical subalgebras and spherical roots
>> https://arxiv.org/pdf/1905.01169v1.pdf
obtain several structure results for a class of spherical subgroups of connected reductive complex algebraic groups that extends the class of strongly solvable spherical subgroups. collect all the necessary material on spherical varieties and provide a detailed presentation of the general strategy for computing the sets of spherical roots.
□ KSHartnett:
Three mathematicians have proven that conducting materials exhibit the ubiquitous statistical pattern known as "universality."
>> https://www.quantamagazine.org/universal-pattern-explains-why-materials-conduct-20190506/

□ GeneSurrounder: network-based identification of disease genes in expression data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2829-y
A more recent category of methods identifies precise gene targets while incorporating systems-level information, but these techniques do not determine whether a gene is a driving source of changes in its network.
The key innovation of GeneSurrounder is the combination of pathway network information with gene expression data to determine the degree to which a gene is a source of dysregulation on the network.

□ Tree reconciliation combined with subsampling improves large scale inference of orthologous group hierarchies
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2828-z
A hierarchy of OGs expands on this notion, connecting more general OGs, distant in time, to more recent, fine-grained OGs, thereby spanning multiple levels of the tree of life.
Large scale inference of OG hierarchies with independently computed taxonomic levels can suffer from inconsistencies between successive levels, such as the position in time of a duplication event.
a new methodology to ensure hierarchical consistency of OGs across taxonomic levels. To resolve an inconsistency, subsample the protein space of the OG members and perform gene tree-species tree reconciliation for each sampling.
□ Single-cell RNA-seq of differentiating iPS cells reveals dynamic genetic effects on gene expression
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/08/630996.full.pdf
with cellular reprogramming becoming an increasingly used tool in molecular medicine, understanding how inter-individual variability effects such differentiations is key.
identify molecular markers that are predictive of differentiation efficiency, and utilise heterogeneity in the genetic background across individuals to map hundreds of eQTL loci that influence expression dynamically during differentiation and across cellular contexts.

□ A new Bayesian methodology for nonlinear model calibration in Computational Systems Biology
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/09/633180.full.pdf
an innovative Bayesian method, called Conditional Robust Calibration (CRC), for nonlinear model calibration and robustness analysis using omics data. CRC is an iterative algorithm based on the sampling of a proposal distributionand on the definition of multiple objective functions, one for each observable.

□ INDRA-IPM: interactive pathway modeling using natural language with automated assembly
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz289/5487381
INDRA (Integrated Network and Dynamical Reasoning Assembler) Interactive Pathway Map (INDRA-IPM), a pathway modeling tool that builds on the capabilities of INDRA to construct and edit pathway maps in natural language and display the results in familiar graphical formats.
INDRA-IPM allows models to be exported in several different standard exchange formats, thereby enabling the use of existing tools for causal inference, visualization and kinetic modeling.
□ mirTime: Identifying Condition-Specific Targets of MicroRNA in Time-series Transcript Data using Gaussian Process Model and Spherical Vector Clustering
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz306/5487390
mirTime uses the Gaussian process regression model to measure data at unobserved or unpaired time points. the clustering performance of spherical k-means clustering for each miRNA when GP was used and when not used, and it was confirmed that the silhouette score was increased.
□ RAxML-NG: A fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz305/5487384
RAxML-NG, a from scratch re-implementation of the established greedy tree search algorithm of RAxML/ExaML
On taxon-rich datasets, RAxML-NG typically finds higher-scoring trees than IQTree, an increasingly popular recent tool for ML-based phylogenetic inference although IQ-Tree shows better stability.

□ Scallop-LR: Quantifying the Benefit Offered by Transcript Assembly on Single-Molecule Long Reads https://www.biorxiv.org/content/biorxiv/early/2019/05/10/632703.full.pdf
Adding long-read-specific algorithms, evolving Scallop to make Scallop-LR, a long-read transcript assembler, to handle the computational challenges arising from long read lengths and high error rates.
Scallop-LR can identify 2100–4000 more known transcripts (in each of 18 human datasets) or 1100–2200 more known transcripts than Iso-Seq Analysis. Further, Scallop-LR assembles 950–3770 more known transcripts and 1.37–2.47 times more potential novel isoforms than StringTie, and has 1.14–1.42 times higher sensitivity than StringTie for the human datasets.
Scallop-LR is a reference-based transcript assembler that follows the standard paradigm of alignment and splice graphs but has a computational formulation dealing with “phasing paths.”
“Phasing paths” are a set of paths that carry the phasing information derived from the reads spanning more than two exons.

□ DeepCirCode: Deep Learning of the Back-splicing Code for Circular RNA Formation
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz382/5488122
DeepCirCode utilizes a convolutional neural network with nucleotide sequence as the input, and shows superior performance over conventional machine learning algorithms such as support vector machine (SVM) and random forest (RF).
Relevant features learnt by DeepCirCode are represented as sequence motifs, some of which match human known motifs involved in RNA splicing, transcription or translation.

□ Exact hypothesis testing for shrinkage based Gaussian Graphical Models
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btz357/5488126
Reconstructing a GGM from data is a challenging task when the sample size is smaller than the number of variables. a proper significance test for the “shrunk” partial correlation (i.e. GGM edges) is an open challenge as a probability density including the shrinkage is unknown.
a geometric reformulation of the shrinkage based GGM, and a probability density that naturally includes the shrinkage parameter. the inference using this new “shrunk” probability density is as accurate as Monte Carlo estimation (an unbiased non-parametric method) for any shrinkage value, while being computationally more efficient.

□ iRNAD: a computational tool for identifying D modification sites in RNA sequence
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz358/5488125
iRNAD is a predictor system for identifying whether a RNA sequence contains D modification sites based on machine learning method.
Support vector machine was utilized to perform classification. The final model could produce the overall accuracy of 96.18% with the area under the receiver operating characteristic curve of 0.9839 in jackknife cross-validation test.
□ Spectrum: Fast density-aware spectral clustering for single and multi-omic data
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/13/636639.full.pdf
Spectrum uses a new density-aware kernel that adapts to data scale and density. It uses a tensor product graph data integration and diffusion technique to reveal underlying structures and reduce noise.
Examining the density-aware kernel in comparison with the Zelnik-Manor kernel demonstrated Spectrum’s emphasis on strengthening local connections in the graph in regions of high density, partially accounts for its performance advantage.
Spectrum is flexible and adapts to the data by using the k-nearest neighbor distance instead of global parameters when performing kernel calculations.
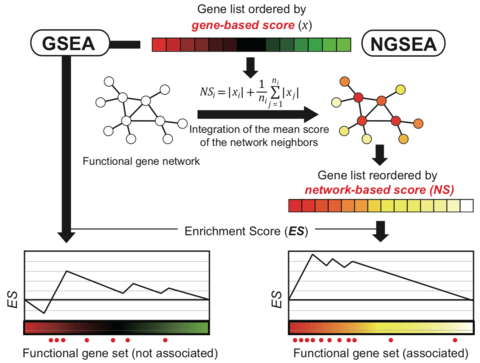
□ NGSEA: network-based gene set enrichment analysis for interpreting gene expression phenotypes with functional gene sets
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/14/636498.full.pdf
network-based GSEA (NGSEA), which measures the enrichment score of functional gene sets using the expression difference of not only individual genes but also their neighbors in the functional network.
NGSEA integrated the mean of the absolute value of the log2(Ratio) for the network neighbors of each gene to account for the regulatory influence on its local subsystem.
□ metaFlye: scalable long-read metagenome assembly using repeat graphs https://www.biorxiv.org/content/biorxiv/early/2019/05/15/637637.full.pdf
metaFlye captures many 16S RNA genes within long contigs, thus providing new opportunities for analyzing the microbial “dark matter of life”.
The Flye algorithm first attempts to approximate the set of genomic k -mers ( k -mers that appear in the genome) by selecting solid k -mers (high-frequency k- mers in the read-set). It further uses solid k-mers to efficiently detect overlapping reads, and greedily combines overlapping reads into disjointigs.
□ A sparse negative binomial classifier with covariate adjustment for RNA-seq data
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/15/636340.full.pdf
Existing methods such as sPLDA (sparse Poisson linear discriminant analysis) does not consider overdispersion properly, NBLDAPE does not embed regularization for feature selection and both methods cannot adjust for covariate effect in gene expression.
a negative binomial model via generalized linear model framework with double regularization for gene and covariate sparsity to accommodate three key elements: adequate modeling of count data with overdispersion, gene selection and adjustment for covariate effect.
□ POLARIS: path of least action analysis on energy landscapes
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/17/633628.full.pdf
POLARIS (Path of Least Action Recursive Survey) provides an alternative approach to the minimum energy pathfinding problem by avoiding the arbitrary assignment of edge weights and extending its methods outside the realm of graph theory.
the algorithm is fully capable of representing the trajectories of highly complex structures within this domain (i.e., the ribosome, contained within a 70×70 dimension landscape).
POLARIS offers the ‘Transition State Weighting’ constraint, which can be enabled to weight the comparison of competing lowest-energy paths based on their rate-limiting step (point of maximal energy through which that path passes) instead of by just the net integrated energy along that path.