









事象全体の複雑性は、一部の高次な複雑性を内包する圏と、
決定論的に作用する余事象の複雑性との総和によって量られる。

□ pathoLogic / plasmIDent: Tracking of antibiotic resistance transfer and rapid plasmid evolution in a hospital setting by Nanopore sequencing
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/17/639609.full.pdf
The sequences of plasmids from multiple isolates of the same or different species are compared in order to identify horizontal gene transfers, structural variations and point mutations, which can further be utilized for phylogenetic or transmission analysis.
the computational platforms pathoLogic and plasmIDent for Nanopore-based characterization of clinical isolates and monitoring of ARG transfer, comprising de-novo assembly of genomes and plasmids, polishing, QC, plasmid circularization, ARG annotation.

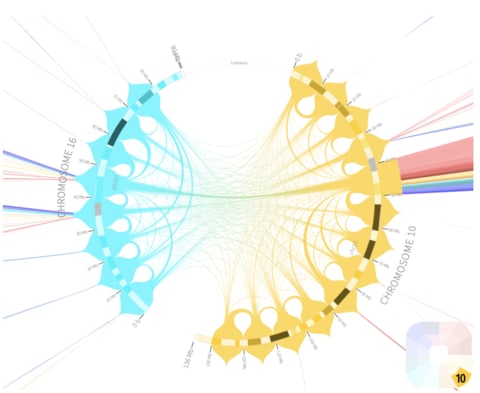
□ Tasks, Techniques, and Tools for Genomic Data Visualization
>> https://arxiv.org/pdf/1905.02853.pdf
As the sequential organization is a key characteristic of genomic data, they limit the scope of this survey to visualizations that incorporate one or more genomic coordinate systems and present data in the order defined by the sequence of that coordinate system.
This explicitly excludes many techniques that are based on reorderable matrices and node-link diagram approaches as matrix-based, clustered heatmaps or visualization of gene regulatory networks as node-link diagrams with expression data mapped overlaid onto the nodes.
□ Insights into the stability of a therapeutic antibody Fab fragment by molecular dynamics and its stabilization by computational design
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/644369.full.pdf
This work elucidated the stability-limiting regions of the antibody fragment Fab A33 using several computational tools,
atomistic molecular dynamics simulations, in-silico mutational analysis by FoldX and Rosetta, packing density calculators, analysis of existing Fab sequences and predictors of aggregation-prone regions.
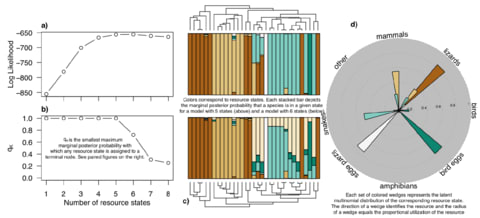
□ Complex ecological phenotypes on phylogenetic trees: a hidden Markov model for comparative analysis of multivariate count data https://www.biorxiv.org/content/biorxiv/early/2019/05/17/640334.full.pdf
Continuous-time Markov chains (CTMC) are commonly used to model ecological niche evolution on phylogenetic trees but are limited by the assumption that taxa are monomorphic and that states are univariate categorical variables.
a hidden Markov model using a Dirichlet-multinomial framework to model resource use evolution on phylogenetic trees. Unlike existing Continuous-time Markov chains (CTMC) implementations, states are unobserved probability distributions from which observed data are sampled.

□ BURMUDA: A novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/17/641191.full.pdf
BERMUDA (Batch-Effect ReMoval Using Deep Autoencoders), a novel transfer-learning-based method for batch-effect correction in scRNA-seq data. BERMUDA can be effectively applied to batches with vastly different cell population compositions, and can properly combine different batches while transferring biological information from one batch to amplify the corresponding signals in other batches.
□ Accuracy, Robustness and Scalability of Dimensionality Reduction Methods for Single Cell RNAseq Analysis
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/17/641142.full.pdf
a comprehensive comparison of different dimensionality reduction methods for scRNAseq analysis based on two important downstream applications: cell clustering and trajectory inference.
Factor models for Dimensionality Reduction is an important modeling part for multiple scRNAseq data sets alignment for integrative analysis of multiple omics data sets, as well as for deconvoluting bulk RNAseq data using cell type specific gene expression measurements from scRNAseq.
the true lineage is linear without any bifurcation or multifurcation patterns, while the inferred lineage may contain multiple ending points in addition to the single starting point. for each inferred lineage, examined one trajectory at a time, where each trajectory consists of the starting point and one of the ending points.
the maximum absolute 𝜏 over all these trajectories as the final Kendall correlation score to evaluate the similarity between the inferred lineage and the true lineage.
□ PhenoGMM: Gaussian mixture modelling of microbial cytometry data enables efficient predictions of biodiversity
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/18/641464.full.pdf
In combination with a supervised machine learning model, diversity estimations based on 16S rRNA gene amplicon sequencing data can be predicted.
PhenoGMM was compared with a generic fixed binning approach called ’PhenoGrid’.
Upon making predictions, PhenoGMM resulted in either more or equally accurate predictions compared to PhenoGrid for all datasets.
Unsupervised estimations of α-diversity resulted in higher correlations with the target diversity values for PhenoGMM for the synthetic communities, while estimations were better for PhenoGrid for natural communities, for which the diversity was determined based on 16S rRNA gene amplicon sequencing.

□ Moment-based Estimation of Mixtures of Regression Models
>> https://arxiv.org/pdf/1905.06467v1.pdf
Using moment-based estimation of the regression parameters, developed the unbiased estimators with a minimum of assumptions on the mixture components.
Finite mixtures of regression models provide a flexible modeling framework for many phenomena.
Zero-inflated regression models, and hurdle models can be considered special cases of the class of finite mixture of regression models with two components.

□ An Information Theoretic Interpretation to Deep Neural Networks
>> https://arxiv.org/pdf/1905.06600v1.pdf
formalize the intuition by showing that the features extracted by DNN coincide with the result of an optimization problem, which we call the
“universal feature selection” problem.
the DNN weight updates in general can be interpreted as projecting features between the feature spaces for extracting the most correlated aspects between them, and the iterative projections can be viewed as computing the SVD of a linear projection between these feature spaces.
□ The free globularily generated double category as a free object
>> https://arxiv.org/pdf/1905.02888v1.pdf
the restriction to the category of globularily generated double categories of the decorated horizontalization functor is faithful. the main ideas behind the free globularily generated double category construction to extend this construction to decorated pseudofunctors.
the free globularily generated double category construction together with the free double functor construction forms a functor from decorated bicategories to globularily generated double categories.
□ Bayesian multivariate reanalysis of large genetic studies identifies many new associations
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/16/638882.full.pdf
the vast majority of GWAS have been analyzed using simple univariate analyses, which consider one phenotype at a time.
Conduct multivariate association analyses on 13 different publicly-available GWAS datasets that involve multiple closely-related phenotypes.
□ The law of genetic privacy: applications, implications, and limitations
>> https://academic.oup.com/jlb/advance-article/doi/10.1093/jlb/lsz007/5489401
the current landscape of genetic privacy to identify the roles that the law does or should play, with a focus on federal statutes and regulations, including the Health Insurance Portability and Accountability Act (HIPAA) and the Genetic Information Nondiscrimination Act (GINA).
□ Multi-insight visualization of multi-omics data via ensemble dimension reduction and tensor factorization
>> https://academic.oup.com/bioinformatics/article-abstract/35/10/1625/5116143
relying only on one single projection can be risky, because it can close our eyes to important parts of the full knowledge space.
The main idea behind the methodology is to combine several Dimension Reduction methods via tensor factorization and group the solutions into an optimal number of clusters.
□ Genotype Imputation and Reference Panel: A Systematic Evaluation
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/19/642546.full.pdf
evaluated the existing reference panels such as the HRC and 1000G Phase3 and CONVERGE.
□ BULQ-Seq: Robust, doublet-free, and low-cost molecular profiling of biological systems
>> https://satijalab.org/img/preprint.pdf
BULQ-seq might ameliorate the extensive false negative (dropouts) associated with scRNA-seq. When examining a scRNA-seq dataset produced on cell lines, the only 1% of the elements in the count matrix were non-zero.
BULQ-seq data exhibited more non-zero values than could be modeled using a standard Zero-Inflated Negative Binomial (ZINB) distribution.

□ Spring Model – chromatin modeling tool based on OpenMM
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/642322.full.pdf
Spring Model (SM) uses OpenMM engine for building models, a fast, simple to use and powerful tool for visualisation of a fiber with a given set of contacts, in 3D space.
the user has to provide contacts and will obtain 3D structure that satisfies these contacts. Additional extra parameters allow controlling fibre stiffness, type of initial structure, resolution. There are also options for structure refinement, and modelling in a spherical container.

□ dearseq: a variance component score test for RNA-Seq differential analysis that effectively controls the false discovery rate
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/635714.full.pdf
dearseq, a new method for DEA which controls the FDR without making any assumption about the true distribution of RNA-seq data. dearseq is a robust approach that uses a variance component score test and relies on nonparametric regression to account for the intrinsic heteroscedasticity of RNA- seq data.
dearseq can efficiently identify the genes whose expression is significantly associated with one or several factors of interest in complex experimental designs (including longitudinal observations) from RNA-seq data while providing robust control of FDR.
□ A general LC/MS-based RNA sequencing method for direct analysis of multiple-base modifications in RNA mixtures
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/643387.full.pdf
The first direct and modification type- independent RNA sequencing method via integration of a hydrophobic end-labeling strategy with of 2-D mass-retention time LC/MS analysis to allow de novo sequencing of RNA mixtures and enhance sample usage efficiency.
This method can directly read out the complete sequence, while identifying, locating, and quantifying base modifications accurately in both single and mixed RNA samples containing multiple different modifications at single-base resolution.
□ Direct prediction of regulatory elements from partial data without imputation
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/643486.full.pdf
an extension to the IDEAS genome segmentation platform which can perform genome segmentation on incomplete regulatory genomics dataset collections without using imputation.
Instead of relying on imputed data, they use an expectation-maximization approach to estimate marginal density functions within each regulatory state.
□ TADdyn: Dynamic simulations of transcriptional control during cell reprogramming reveal spatial chromatin caging
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/642009.full.pdf
Chromosome Conformation Capture (3C)-based experiments combined with computational modelling are pivotal for unveiling 3D chromosome structure.
TADdyn, a new tool that integrates time-course 3C data, restraint-based modelling, and molecular dynamics to simulate the structural rearrangements of genomic loci in a completely data-driven way.
□ Inducible ANT RNA-Seq:
>> https://bitbucket.org/lorainelab/inducible-ant-rna-seq/src/master/
This project analyzes RNA-Seq data from inducing ANT expression over a time course. Goal is to identify direct targets of ANT regulation.
□ Robinson-Foulds Reticulation Networks
>> https://www.biorxiv.org/content/10.1101/642793v1
Given a collection of phylogenetic input trees, this problem seeks a minimum reticulation network with the smallest number of reticulation vertices into which the input trees can be embedded exactly.
Unfortunately, this problem is limited in practice, since minimum reticulation networks can be easily obfuscated by even small topological errors that typically occur in input trees inferred from biological data.
The adapted problem, called the Robinson-Foulds reticulation network (RF-Network) problem is, as we show and like many other problems applied in molecular biology, NP-hard.

□ An omnidirectional visualization model of personalized gene regulatory networks
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/644070.full.pdf
a generalized framework for inferring informative, dynamic, omnidirectional, and personalized GRNs (idopGRNs) from routine transcriptional experiments. This framework is constructed by a system of quasi-dynamic ordinary differential equations (qdODEs) derived from the combination of ecological and evolutionary theories.
□ Retroposon Insertions within a Multispecies Coalescent Framework Suggest that Ratite Phylogeny is not in the 'Anomaly Zone'
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/643296.full.pdf
The MP-EST species tree suggests an empirical case of the 'anomaly zone' with three very short internal branches at the base of Palaeognathae, and as predicted for anomaly zone conditions, the MP-EST species tree differs from the most common gene tree.
ASTRAL is used to estimate a species tree in the statistically consistent framework of the multispecies coalescent. Although identical in topology to the MP-EST tree, the ASTRAL species tree based on retroposons shows branch lengths that are much longer and incompatible with anomaly zone conditions.
□ Do signaling networks and whole-transcriptome gene expression profiles orchestrate the same symphony?
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/643866.full.pdf
using four common and comprehensive databases i.e. GEO, GDSC, KEGG, and OmniPath, extracted all relevant gene expression data and all relationships among directly linked gene pairs in order to evaluate the rate of coherency or sign consistency.
the ratios for the analysis based on OmniPath and GDSC is more uniformly distributed hold a candle to others and there is not any kind of dual feedback loop structures i.e. DNFBL and DPFBLs in OmniPath signaling network which can be controversy.
Most of these kinds of altred expression are disappeared gradually and ignored by the whole system of signaling network either stimulated endogenously or exogenously.

□ Algorithmic differentiation improves the computational efficiency of OpenSim-based optimal control simulations of movement
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/21/644245.full.pdf
an interface between OpenSim and CasADi to perform optimal control simulations.
an alternative to finite differences (FD) for evaluating the derivative matrices required by the NLP solver, namely the objective function gradient, the constraint Jacobian, and the Hessian of the Lagrangian (henceforth referred to as simply Hessian).

□ Needlestack: an ultra-sensitive variant caller for multi-sample next generation sequencing data
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/21/639377.full.pdf
Needlestack, a highly sensitive variant caller, which directly learns from the data the level of systematic sequencing errors to accurately call mutations. Needlestack is based on the idea that the sequencing error rate can be dynamically estimated from analyzing multiple samples together.
Needlestack provides a multi-sample VCF file containing all candidate variants that obtain a QVAL higher than the input threshold in at least one sample, general information about the variant in the INFO field and individual information in the GENOTYPE field.
□ bwa-mem2 pre-release.
>> https://github.com/bwa-mem2/bwa-mem2/compare/v2.0pre1...master
“Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems” (M. Vasimuddin, Sanchit Misra, Heng Li, Srinivas Aluru) Bwa-mem2 paper soon to be open-sourced.
Identical alignments. 80% faster. Other optimizations include software prefetching, removed suffix array compression (higher memory), AVX2/512 instructions e.g. in banded Smith-Waterman.
bwa-mem2 detects the underlying harware flags for AVX512/AVX2/SSE2 vector modes and compiles accordingly. If the platform does not support any AVX/SSE vector mode then it compiles the code in fully scalar mode.
□ Trepli-ATAC-seq: ranscription Restart Establishes Chromatin Accessibility after DNA Replication
>> https://www.cell.com/molecular-cell/fulltext/S1097-2765(19)30352-1
Chromatin accessibility restores differentially genome wide, with super enhancers regaining transcription factor occupancy faster than other genomic features.
Systematic inhibition of transcription shows that transcription restart is required to re-establish active chromatin states genome wide and resolve opportunistic binding events resulting from DNA replication.
□ CAS: Context-Aware Seeds for Read Mapping
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/20/643072.full.pdf
CAS guarantees finding all valid mapping but uses fewer (and longer) seeds, which reduces seed frequencies and increases efficiency of mappers.
CAS generalizes the existing pigeonhole-principle-based seeding scheme in which this confidence radius is implicitly always 1, and design an efficient algorithm that constructs the confidence radius database in linear time.
□ Estimating the Strength of Expression Conservation from High Throughput RNA-seq Data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz405/5494699
a gamma distribution model to describe how the strength of expression conservation (denoted by W) varies among genes.
Given the high throughput RNA-seq datasets from multiple species, we then formulate an empirical Bayesian procedure to estimate W for each gene. those W-estimates are useful to study the evolutionary pattern of expression conservation.

□ FROM MANHATTAN PLOT TO BIGTOP: DNANEXUS MAKES DATA VISUALIZATION A (VIRTUAL) REALITY
>> https://blog.dnanexus.com/2019-05-21-bigtop-data-visualization/
While BigTop is meant for examining large genomic data sets such as those found in GWAS studies, in reality it can be used to visualize any data set that contains genomic location, p-value, and a factor that can be quantified between 0 and 1.
□ Large time asymptotics for a cubic nonlinear Schrödinger system in one space dimension
>> https://arxiv.org/pdf/1905.07123v1.pdf
a two-component system of cubic nonlinear Schro ̈dinger equations in one space dimension.
The each component of the solutions to this system behaves like a free solution in the large time, but there is a strong restriction between the profiles of them. This turns out to be a consequence of non-trivial long-range nonlinear interactions.
□ Linear time minimum segmentation enables scalable founder reconstruction
>> https://almob.biomedcentral.com/articles/10.1186/s13015-019-0147-6
Given a minimum segment length and m sequences of length n drawn from an alphabet of size σ, create a segmentation in O(mn log σ) time and use various matching strategies to join the segment texts to generate founder sequences.
Optimizing the founder set is an NP-hard problem, but there is a segmentation formulation that can be solved in polynomial time. an O(mn) time (i.e. linear time in the input size) algorithm to solve the minimum segmentation problem for founder reconstruction, improving over an earlier O(mn^2).

□ C-InterSecture – a computational tool for interspecies comparison of genome architecture
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz415/5497251
C-InterSecture, a computational pipeline allowing systematic comparison of genome architecture between species.
C-InterSecture allows statistical comparison of contact frequencies of individual pairs of loci, as well as interspecies comparison of contacts pattern within defined genomic regions, i.e. topologically associated domains.
C-InterSecture was designed to liftover contacts between species, compare 3-dimensional organization of defined genomic regions, such as TADs, and analyze statistically individual contact frequencies.
□ SPar-K : a method to partition NGS signal data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz416/5497248
SPar-K (Signal Partitioning using K-means) is a modified version of a standard K-means algorithm designed to cluster vectors containing a sequence of signal (that is, the order in which the elements appear in the vectors is meaningful).
This method efficiently deals with problems of data heterogeneity, limited misalignment of anchor points and unknown orientation of asymmetric patterns.
In order to detect a possible phase shift or orientation inversion between two vectors, this program allows computing distances between two vectors by shifting and flipping them.
□ PUMILIO, but not RBMX, binding is required for regulation of genomic stability by noncoding RNA NORAD
>> https://www.biorxiv.org/content/biorxiv/early/2019/05/22/645960.full.pdf
addressing the relative contributions of NORAD:PUM and NORAD:RBMX interactions to the regulation of genomic stability by this lncRNA.
Extensive RNA FISH and fractionation experiments established that NORAD localizes predominantly to the cytoplasm with or without DNA damage.
genetic rescue experiments demonstrated that PUM binding is required for maintenance of genomic stability by NORAD whereas binding of RBMX is dispensable for this function.
These data therefore establish an essential role for the NORAD:PUM interaction in genome maintenance and provide a foundation for further mechanistic dissection of this pathway.
□ SCTree: Statistical test of structured continuous trees based on discordance matrix
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz425/5497258
SCTree test is an algorithm that can statistical detect the hidden structure of high-dimensional single-cell dataset, which the intrinsic structure may be linear structure of branched structure.
Based on the tools of spiked matrix model and random matrix theory, SCTree construct the discordance matrix by transforming the distance between any pair of cells used Gromov-Farris transform.
□ simuG: a general-purpose genome simulator
>> https://github.com/yjx1217/simuG
Simulated genomes with pre-defined or random genomic variants can be very useful for benchmarking genomic and bioinformatics analyses. simuG as a light-weighted tool for simulating the full spectrum of genomic variants (SNPs, INDELs, CNVs, inversions, translocations).
simuG enables a rich array of fine-tuned controls, such as simulating SNPs in different coding partitions (e.g. coding sites, noncoding sites, 4-fold degenerate sites, or 2-fold degenerate sites);
simulating CNVs with different formation mechanisms (e.g. segmental deletions, dispersed duplications, and tandem duplications); and simulating inversions and translocations with specific types of breakpoints.
□ SureTypeSC - A Random Forest and Gaussian Mixture predictor of high confidence genotypes in single cell data
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btz412/5497252
SureTypeSC - a two-stage machine learning algorithm that filters a substantial part of the noise, thereby retaining the majority of the high quality SNPs. SureTypeSC also provides a simple statistical output to show the confidence of a particular single cell genotype using Bayesian statistics.
SureTypeSC is implementation of algorithm for regenotyping of single cell data coming from Illumina BeadArrays.
