□ PΨFinder: a practical tool for the identification and visualization of novel pseudogenes in DNA sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04583-4
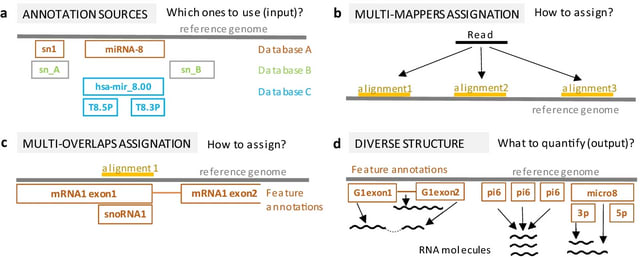
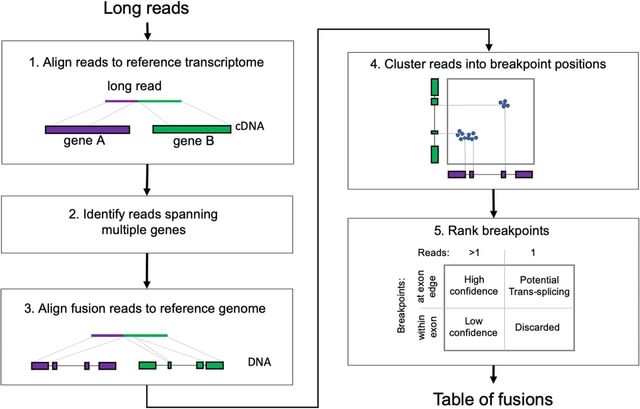
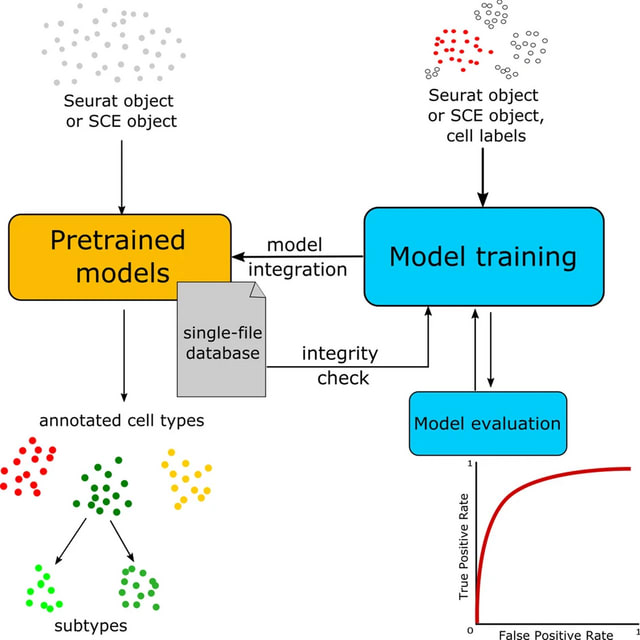
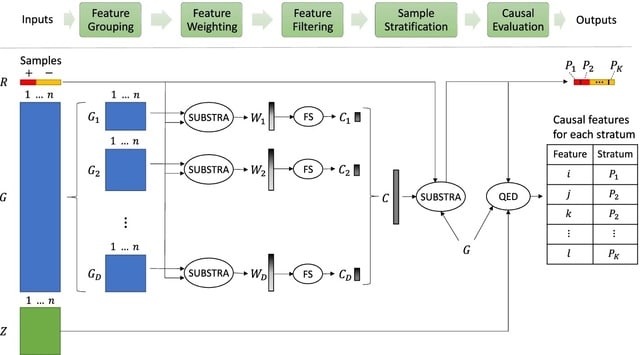
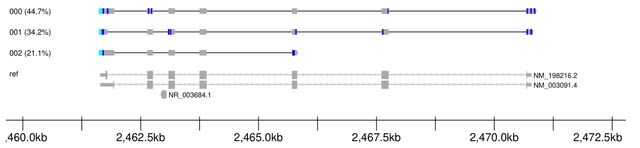
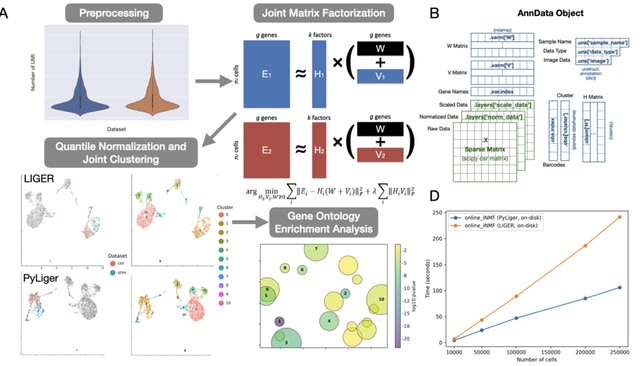
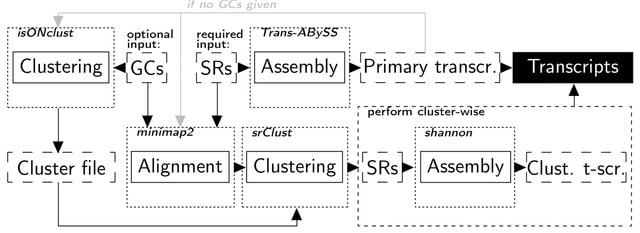
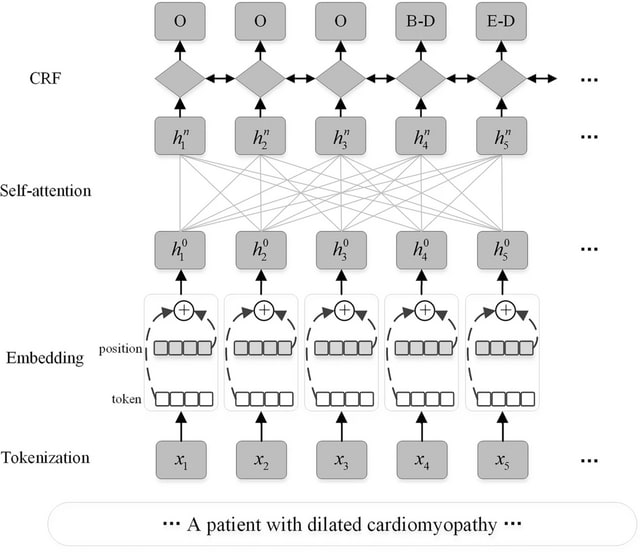
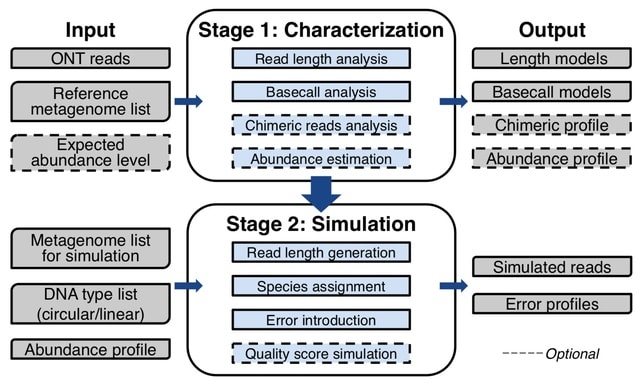
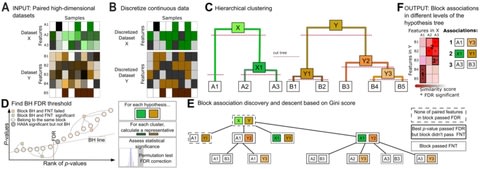
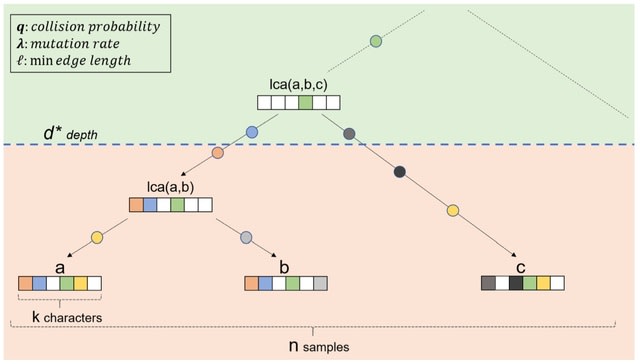
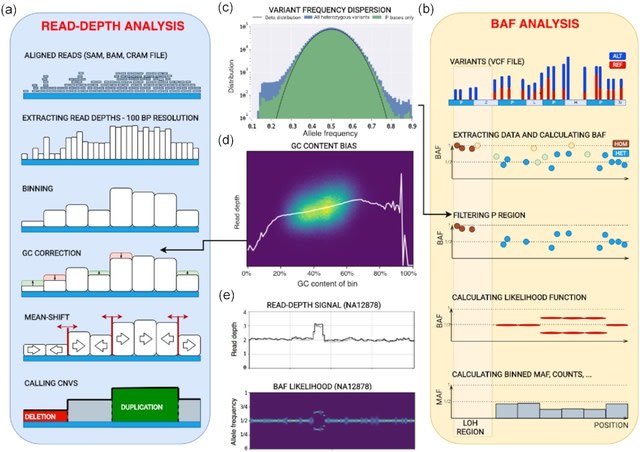
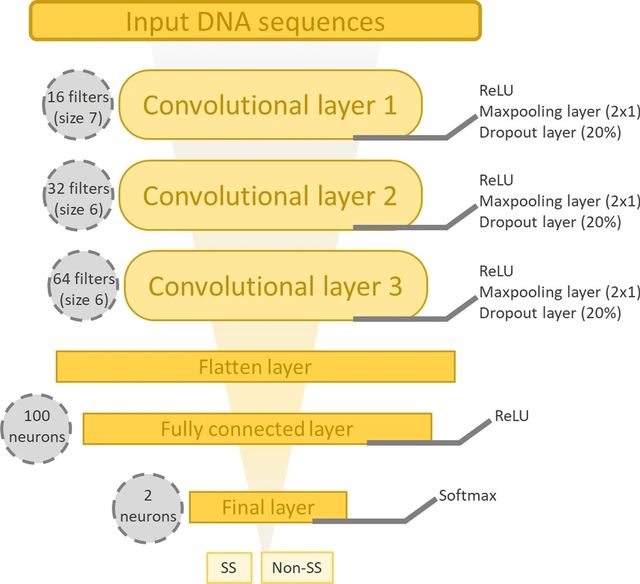
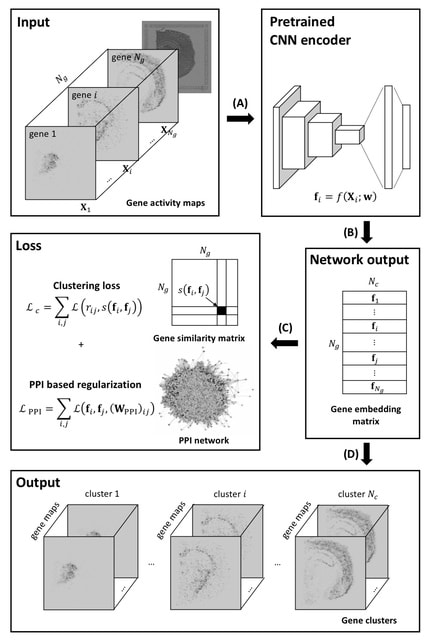
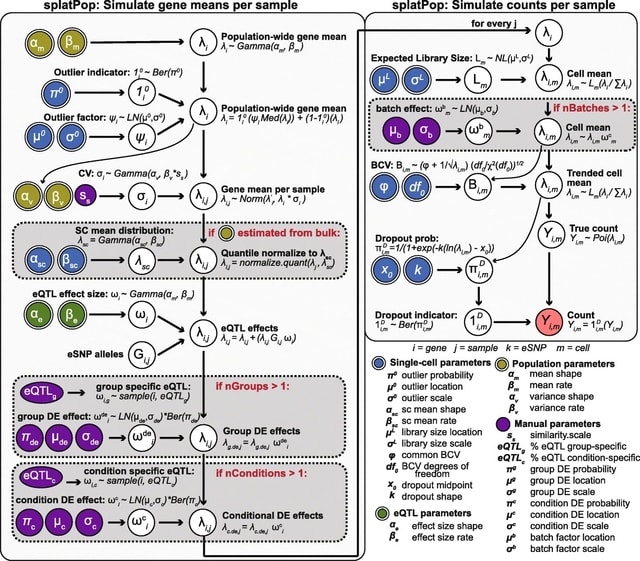
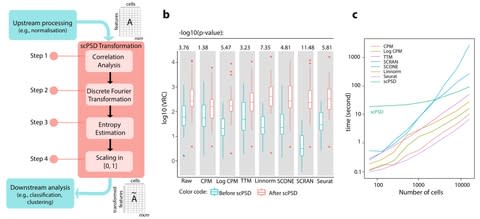
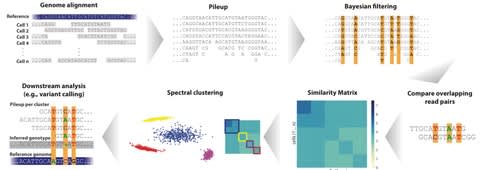
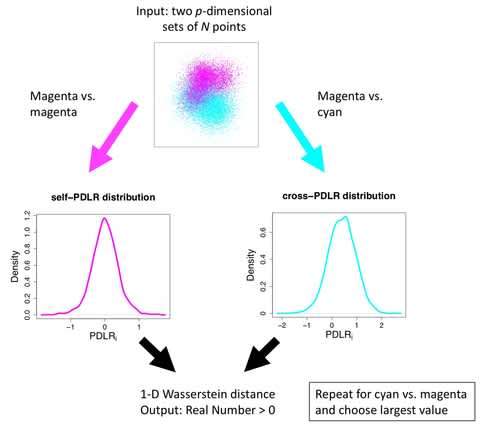
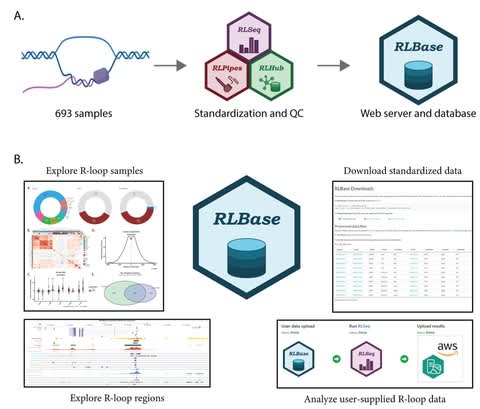
PΨFinder can automatically identify novel PΨgs from DNA sequencing data and determine their location in the genome with high sensitivity. Insert positions of the pseudogene candidates are recorded by linking the pseudogene candidate with chimeric reads and chimeric pairs.
The resulting analysis with PΨFinder, determined that predictions obtained from samples with a sequencing depth of 5 M reads, an average coverage of at least 144X and including both CPs and CRs, can be deemed as true positive PΨg-insertion sites.

□ A scalable and unbiased discordance metric with H+
>> https://www.biorxiv.org/content/10.1101/2022.02.03.479015v1.full.pdf
H+, a modification of G+ that retains the scale-agnostic discordance quantification while addressing problems with G+. Explicitly, H+ is an unbiased estimator for P (dij ) > P (dkl ).
An estimate of H+ based on bootstrap resampling from the original observations that does not require the full dissimilarity matrices to be calculated. H+ provides an additional means to consider termination of a clustering algorithm in a distance-agnostic manner.

□ Omics-informed CNV calls reduce false positive rate and improve power for CNV-trait associations
>> https://www.biorxiv.org/content/10.1101/2022.02.07.479374v1.full.pdf
A method to improve the detection of false positive CNV calls amongst PennCNV output by discriminating between high quality (true) and low quality (false) CNV regions based on multi-omics data.
a predictor of CNV quality inferred from WGS, transcriptomics and methylomics, solely based on PennCNV software output parameters in these samples assayed by multiple omics technologies.

□ scHFC: a hybrid fuzzy clustering method for single-cell RNA-seq data optimized by natural computation
>> https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab588/6523126
scHFC is a hybrid fuzzy clustering method optimized by natural computation based on Fuzzy C Mean (FCM) and Gath-Geva (GG) algorithms. Specifically, principal component analysis algorithm is utilized to reduce the dimensions of scRNA-seq data after it is preprocessed.
FCM algorithm optimized by simulated annealing algorithm and genetic algorithm is applied to cluster the data to output a membership matrix, which represents the initial clustering result and is taken as the input for GG algorithm to get the final clustering results.
a cluster number estimation method called multi-index comprehensive estimation, which can estimate the cluster numbers well by combining four clustering effectiveness indexes.

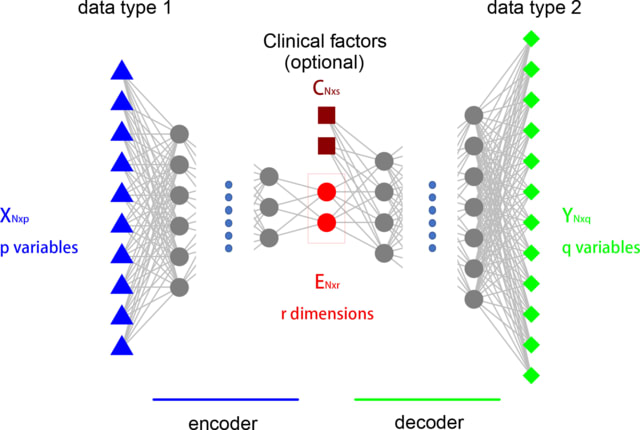
□ expiMap: Biologically informed deep learning to infer gene program activity in single cells
>> https://www.biorxiv.org/content/10.1101/2022.02.05.479217v1.full.pdf
The key concept is the substitution of the uninterpretable nodes in an autoencoder’s bottleneck by labeled nodes mapping to interpretable lists of genes, such as gene ontologies, or biological pathways, for which activities are learned as constraints during reconstruction.
expiMap, “explainable programmable mapper” is consist of interpretable CVAE that allows the incorporation of domain knowledge by “architecture programming”, i.e., constraining the network architecture to ensure that each latent dimension captures the variability of known GPs.

□ Changes in chromatin accessibility are not concordant with transcriptional changes for single-factor perturbations
>> https://www.biorxiv.org/content/10.1101/2022.02.03.478981v1.full.pdf
Integrating tandem, genome-wide chromatin accessibility and transcriptomic data to characterize the extent of concordance between them in response to inductive signals.
While certain genes have a high degree of concordance of change between expression and accessibility changes, there is also a large group of differentially expressed genes whose local chromatin remains unchanged.

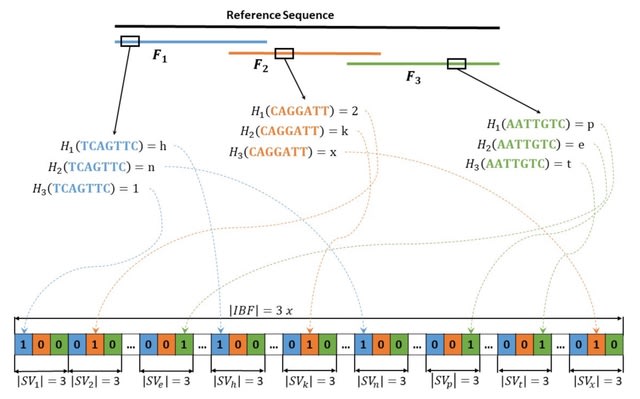
□ StructuralVariantAnnotation: a R/Bioconductor foundation for a caller-agnostic structural variant software ecosystem
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac042/6522107
StructuralVariantAnnotation provides the caller-agnostic foundation needed for a R/Bioconductor ecosystem of structural variant annotation, classification, and interpretation tools able to handle both simple and complex genomic rearrangements.
StructuralVariantAnnotation can match equivalent variants reported as insertion and duplication and can identify transitive breakpoints. Such features are important as they are common when comparing short and long read call sets.

□ SAMAR: Assembly-free rapid differential gene expression analysis in non-model organisms using DNA-protein alignment
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-08278-7
SAMAR (Speedy, Assembly-free Method to Analyze RNA-seq expression data) -- a quick-and-easy way to perform differential expression (DE) analysis in non-model organisms.
SAMAR uses LAST to learn the alignment scoring parameters suitable for the input, and to estimate the paired-end fragment size distribution of paired-end reads, and directly align RNA-seq reads to the high-confidence proteome that would have been otherwise used for annotation.

□ Fast and compact matching statistics analytics
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac064/6522115
A parallel algorithm for shared-memory machines that computes matching statistics 30 times faster with 48 cores in the cases that are most difficult to parallelize.
A a lossy compression scheme that shrinks the matching statistics array to a bitvector that takes from 0.8 to 0.2 bits per character, depending on the dataset and on the value of a threshold, and that achieves 0.04 bits per character in some variants.
Efficient implementations of range-maximum and range-sum queries that take a few tens of milliseconds while operating on our compact representations, and that allow computing key local statistics about the similarity between two strings.

□ FUNKI: Interactive functional footprint-based analysis of omics data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac055/6522117
FUNKI, a FUNctional toolKIt for footprint analysis. It provides a user-friendly interface for an easy and fast analysis of transcriptomics, phosphoproteomics and metabolomics data, either from bulk or single-cell experiments.
FUNKI provides a user interface to upload omics data, and then run DoRothEA, PROGENy, KinAct, CARNIVAL and COSMOS to estimate the activity of pathways, transcription factors, and kinases. The results are visualized in diverse forms.

□ CRFalign: A Sequence-structure alignment of proteins based on a combination of HMM-HMM comparison and conditional random fields
>> https://www.biorxiv.org/content/10.1101/2022.02.03.478675v1.full.pdf
CRFalign improves upon a reduced three-state or five-state scheme of HMM-HMM profile alignment model by means of conditional random fields with nonlinear scoring on sequence and structural features implemented with boosted regression trees.
CRFalign extracts complex nonlinear relationships among sequence profiles & structural features incl secondary structures/solvent accessibilities/environment-dependent properties that give rise to position-dependent as well as environment-dependent match scores and gap penalties.

□ Beware to ignore the rare: how imputing zero-values can improve the quality of 16S rRNA gene studies results
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04587-0
A collection of normalization and zero-imputation approaches is tested for 16S rRNA-gene sequencing data preprocessing. This permits to compare an updated list of normalization tools considering the recent publications and evaluate the effect of introducing zero-imputation step.
Bray–Curtis dissimilarity was used to build a distance matrix on which Non-metric Multidimensional Scaling (NMDS) dimensionality reduction was performed to assess spatial distribution of samples, whereas Whittaker dissimilarity values were graphically represented using heatmaps.

□ Belayer: Modeling discrete and continuous spatial variation in gene expression from spatially resolved transcriptomics
>> https://www.biorxiv.org/content/10.1101/2022.02.05.479261v1.full.pdf
In the simplest case of an axis-aligned tissue structure, Belayer infers the maximum likelihood expression function using a dynamic programming algorithm that is related to the classical problems of changepoint detection and segmented regression.
Belayer models the expression of each gene with a piecewise linear expression function. And analyzes spatially resolved transcriptomics data using a global model of tissue organization and an explicit definition of GE that combines both discrete and continuous variation in space.

□ SpatialCorr: Identifying Gene Sets with Spatially Varying Correlation Structure
>> https://www.biorxiv.org/content/10.1101/2022.02.04.479191v1.full.pdf
SpatialCorr, a semiparametric approach for identifying spatial changes in the correlation structure of a group of genes. SpatialCorr estimates spot-specific correlation matrices using a Gaussian kernel; region-specific correlations are estimated using all spots in a region.
SpatialCorr tests for spatially varying correlation within each tissue region using a multivariate normal (MVN) likelihood ratio test statistic that compares the MVN w/ spot-specific correlation estimates to an MVN with constant correlation estimated from all spots in the region.

□ CRSP: Comparative RNA-seq pipeline for species lacking both of sequenced genomes and reference transcripts
>> https://www.biorxiv.org/content/10.1101/2022.02.04.479193v1.full.pdf
CRSP integrates a set of computational strategies, such as mapping de novo transcriptomic assembly contigs to a protein database, using Expectation-Maximization (EM) algorithm to assign reads mapping uncertainty, and integrative statistics to quantify gene expression values.
CRSP estimated gene expression values are highly correlated with gene expression values estimated by directly mapping to a reference genome.
10 to 20 million single-end reads are sufficient to achieve reasonable gene expression quantification accuracy while a pre-compiled de novo transcripts assembly from deep sequencing can dramatically decrease the minimal reads requirement for the rest of RNA-seq experiments.

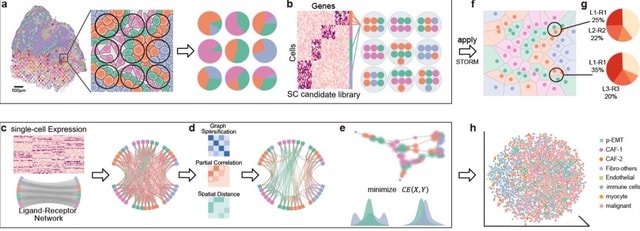
□ LRLoop: Feedback loops as a design principle of cell-cell communication
>> https://www.biorxiv.org/content/10.1101/2022.02.04.479174v1.full.pdf
Currently available techniques for predicting ligand-receptor interactions are one-directional from sender to receiver cells.
LRLoop, a new method for analyzing cell-cell communication that is based on bi-directional ligand-receptor interactions, where two pairs of ligand-receptor interactions are identified that are responsive to each other, and thereby form a closed feedback loop.

□ Thirdkind: displaying phylogenetic encounters beyond 2-level reconciliation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac062/6525213
No simple generic tool is available to visualise reconciliation results. Moreover there is no tool to visualise 3-levels reconciliations, i.e. to visualise 2 nested reconciliations as for example in a host/symbiont/gene complex.
Thirdkind is a light command-line software allowing the user to generate a svg from recPhyloXML files with a large choice of options (orientation, police size, branch length, multiple trees, redundant transfers handling, etc.) and to handle the visualisation of 2 nested reconciliations.

□ SPRI: Spatial Pattern Recognition using Information based method for spatial gene expression data
>> https://www.biorxiv.org/content/10.1101/2022.02.09.479510v1.full.pdf
SPRI directly models spatial transcriptome raw count data without model assumptions, which transforms the problem of spatial expression pattern recognition into the detection of dependencies between spatial coordinate pairs with gene read count as the observed frequencies.
SPRI converts the spatial gene pattern problem into an association detection problem b/n coordinate values with observed raw count data, and then estimates associations using an information-based method, TIC, which calculates the total mutual information with all possible grids.

□ blitzGSEA: Efficient computation of Gene Set Enrichment Analysis through Gamma distribution approximation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac076/6526383
blitzGSEA, an algorithm that is based on the same running sum statistic as GSEA, but instead of performing permutations, blitzGSEA approximates the enrichment score probabilities based on Gamma distributions.
The blitzGSEA calculates a background distribution analytically for the weighted Kolmogorov-Smirnov statistic described in GSEA-P and fGSEA using the gene set shuffling methodology.

□ SCAR: Recombination-aware Phylogeographic Inference Using the Structured Coalescent with Ancestral Recombination
>> https://www.biorxiv.org/content/10.1101/2022.02.08.479599v1.full.pdf
the Structured Coalescent with Ancestral Recombination (SCAR) model, which builds on recent approximations to the structured coalescent by incorporating recombination into the ancestry of sampled individuals.
The SCAR model allows us to infer how the migration history of sampled individuals varies across the genome from ARGs, and improves estimation of key population genetic parameters. SCAR explores the potential and limitations of phylogeographic inference using full ARGs.

□ iDESC: Identifying differential expression in single-cell RNA sequencing data with multiple subjects
>> https://www.biorxiv.org/content/10.1101/2022.02.07.479293v1.full.pdf
A zero-inflated negative binomial mixed model is used to consider both subject effect and dropouts. iDESC models dropout events as inflated zeros and non-dropout events using a negative binomial distribution.
In the negative binomial component, a random effect is used to separate subject effect from group effect. Wald statistic is used to assess the significance of group effect.

□ Efficient Privacy-Preserving Whole Genome Variant Queries
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac070/6527622
This project provides a method that uses secure multi-party computation (MPC) to query genomic databases in a privacy-protected manner.
The proposed solution privately outsources genomic data from arbitrarily many sources to the two non-colluding proxies and allows genomic databases to be safely stored in semi-honest cloud. It provides data privacy, query privacy, and output privacy by using XOR-based sharing.
It is possible to query a genomic database with 3, 000, 000 variants with five genomic query predicates under 400 ms. Querying 1, 048, 576 genomes, each containing 1, 000, 000 variants, for the presence of five different query variants can be achieved approximately in 6 minutes.

□ PAC: Scalable sequence database search using Partitioned Aggregated Bloom Comb-Trees
>> https://www.biorxiv.org/content/10.1101/2022.02.11.480089v1.full.pdf
PAC index construction works in a streaming fashion without any disk footprint besides the index itself. It shows a 3 to 6 fold improvement in construction time compared to other compressed methods for comparable index size.
Using inverted indexes and a novel data structure dubbed aggregative Bloom filters, a PAC query can need single random access and be performed in constant time in favorable instances.
As in SBTs, these trees’ inner nodes are unions of Bloom filters, but for PAC, they are organized in a binary left-comb tree and another binary Bloom right-comb tree. Each aggregative Bloom comb tree indexes all k-mers sharing a given minimizer.

□ Degeneracy measures in biologically plausible random Boolean networks
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04601-5
Although degeneracy is a feature of network topologies and seems to be implicated in a wide variety of biological processes, research on degeneracy in biological networks is mostly limited to weighted networks.
An information theoretic definition of degeneracy on random Boolean networks. Random Boolean networks are discrete dynamical systems with binary connectivity and thus, these networks are well-suited for tracing information flow and the causal effects.

□ BinSPreader: refine binning results for fuller MAG reconstruction
>> https://www.biorxiv.org/content/10.1101/2022.02.14.480326v1.full.pdf
BinSPreader — a novel binning refiner tool that exploits the assembly graph topology and other connectivity information to refine the existing binning, correct binning errors, propagate binning from longer contigs to shorter contigs and infer contigs belonging to multiple bins.
BinSPreader can split input reads in accordance with the resulting binning, predicting reads potentially belonging to multiple MAGs.
BinSPreader uses a special working mode of the binning refining algorithm for sparse binnings, where the total length of initially binned contigs is significantly lower than the total assembly length.

□ scShapes: A statistical framework for identifying distribution shapes in single-cell RNA-sequencing data
>> https://www.biorxiv.org/content/10.1101/2022.02.13.480299v1.full.pdf
While most methods for differential gene expression analysis aim to detect a shift in the mean of expressed values, single cell data are driven by over-dispersion and dropouts requiring statistical distributions that can handle the excess zeros.
scShapes quantifies cell-to-cell variability by testing for differences in the expression distribution while flexibly adjusting for covariates. scShapes identifies subtle variations that are independent of altered mean expression and detects biologically-relevant genes.

□ gcaPDA: a haplotype-resolved diploid assembler
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04591-4
gcaPDA (gamete cells assisted Phased Diploid Assembler) can generate chromosome-scale phased diploid assemblies for highly heterozygous and repetitive genomes using PacBio HiFi data, Hi-C data and gamete cell WGS data.
Both of the reconstructed haplotype assemblies generated using gcaPDA have excellent collinearity with their corresponding reference assemblies.
gcaPDA used all the HiFi reads to construct assembly graphs, with haplotype-specific k-mer derived from gamete cell reads to assist in resolving graph, and generate both haplotype assembly simultaneously.

□ Efficient Bayesian inference for mechanistic modelling with high-throughput data
>> https://www.biorxiv.org/content/10.1101/2022.02.14.480336v1.full.pdf
Inspired by the method of stochastic gradient descent (SGD) in machine learning, a minibatch approach tackles this issue: for each comparison between simulated and observed data, it uses a stochastically sampled subset (minibatch) of the data.
Choosing a large enough minibatch ensures that the relevant signatures in the observed data can be accurately estimated, while avoiding unnecessary comparisons that slow down inference.

□ GMMchi: Gene Expression Clustering Using Gaussian Mixture Modeling
>> https://www.biorxiv.org/content/10.1101/2022.02.14.480329v1.full.pdf
Since the iterative process within GMMchi is based on the chi-square goodness of fit test as the main criterion for measuring the fit of the mixed normal distribution model, it is important for the validity of the Chi-square test to have at least 5 measurements in each bin.
This is achieved by an algorithm called dynamic binning, which involves automatically combining bins while applying the least manipulation to the histogram for ensuring optimal results of the underlying chi-square test within GMMchi.

□ CNGPLD: Case-control copy-number analysis using Gaussian process latent difference
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac096/6530274
CNGPLD is a new tool for performing case-control somatic copy-number analysis that facilitates the discovery of differentially amplified or deleted copy-number aberrations in a case group of cancer compared to a control group of cancer.
This tool uses a Gaussian process statistical framework in order to account for the covariance structure of copy-number data along genomic coordinates and to control the false discovery rate at the region level.
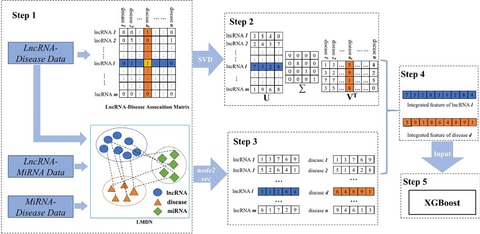
□ DeepMNE: Deep Multi-network Embedding for lncRNA-Disease Association prediction
>> https://ieeexplore.ieee.org/document/9716828/
DeepMNE discovers potential lncRNA disease associations, especially for novel diseases and lncRNAs. DeepMNE extracts multi-omics data to describe diseases and lncRNAs, and proposes a network fusion method based on deep learning to integrate multi-source information.
DeepMNE complements the sparse association network and uses kernel neighborhood similarity to construct disease similarity and lncRNA similarity networks. DeepMNE also elicits a considerable predictive performance on perturbed datasets.
□ Penguin: A Tool for Predicting Pseudouridine Sites in Direct RNA Nanopore Sequencing Data
>> https://www.sciencedirect.com/science/article/abs/pii/S1046202322000354
Penguin integrates several Machine learning models (i.e., predictors) to identify RNA Ψ sites in Nanopore direct RNA sequencing reads. Penguin extracts a set of features from the raw signal measured by the Oxford Nanopore and the corresponding basecalled k-mer.
Penguin automates the data preprocessing incl. Nanopore direct RNA read alignment using Minimap2, and Signal extraction using Nanopolish, feature extraction from raw Nanopore signal for ML predictors integrated, and the prediction of RNA Ψ sites with those predictors.

□ SWIF(r): Enabling interpretable machine learning for biological data with reliability scores
>> https://www.biorxiv.org/content/10.1101/2022.02.18.481082v1.full.pdf
SWIF(r) (SWeep Inference Framework (controlling for correlation)), a supervised machine learning algorithm that applied to the problem of identifying genomic sites under selection in population genetic data. SWIF(r) learns the individual and joint distributions of attributes.
SWIF(r)’s algorithm classifies testing data according to these distributions along with user-provided priors on the relative frequencies of the classes. the SWIF(r) probabilities reported do not offer insight into the degree of "trustworthiness" of a particular classified instance.