










□ SELINA: Single-cell Assignment using Multiple-Adversarial Domain Adaptation Network with Large-scale References
>> https://www.biorxiv.org/content/10.1101/2022.01.14.476306v1.full.pdf
SELINA (single cELl identity NAvigator) optimizes the annotation for minority cell types by synthetic minority over-sampling, removes batch effects using a multiple-adversarial domain adaptation network (MADA), and fits the query data with reference data using an autoencoder.
SELINA affords a comprehensive and uniform reference atlas with 1.7 million cells covering 230 major human cell types.
SELINA multiplies its gene expression vector by a random weight and then sums the pair of weighted vectors to obtain a synthetic cell which is at a random point on the line connecting the pair of cells. SELINA freezes the decoder and turn to update the parameters of the encoder.
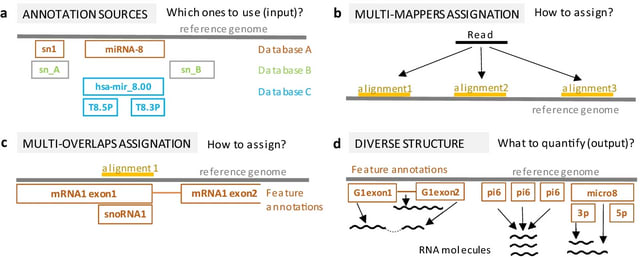
□ MGcount: a total RNA-seq quantification tool to address multi-mapping and multi-overlapping alignments ambiguity in non-coding transcripts
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04544-3
Multi-Graph count (MGcount) assigns reads hierarchically to small-RNA and long-RNA features to account for length disparity when transcripts overlap in the same genomic position.
MGcount outputs a transcriptomic count matrix compatible with RNA-sequencing downstream analysis pipelines, with both bulk and single-cell resolution, and the graphs that model repeated transcript structures for different biotypes.
MGcount aggregates RNA products with similar sequences where reads systematically multi-map using a graph-based approach. The map equation formulates the theoretical limit to compress the description of an infinite random walk trajectory.

□ LDA: Supervised dimensionality reduction for exploration of single-cell data by Hybrid Subset Selection - Linear Discriminant Analysis
>> https://www.biorxiv.org/content/10.1101/2022.01.06.475279v1.full.pdf
LDA (linear discriminant analysis) identifies linear combinations of predictors that optimally separate a priori classes, enabling users to tailor visualizations to separate specific aspects of cellular heterogeneity.
Hybrid-Subset-Selection - LDA performs feature selection to enhance dimensionality reduction and visualization of single-cell data by maximizing class separation via a stepwise feature selection approach, selecting the final model based on a separation metric.
□ NetSeekR: a network analysis pipeline for RNA-Seq time series data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04554-1
an integration of one of the best performing spliced aligners—STAR—with a pseudo-aligner—Kallisto—as well as two differential gene expression analysis tools (edgeR and Sleuth) using different statistical models and data analysis and visualization methods.
NetSeekR, an RNA-Seq data analysis R package aimed at analyzing the transcriptome dynamics for inferring networks of differentially expressed genes associated with experimental treatments measured at multiple time points.
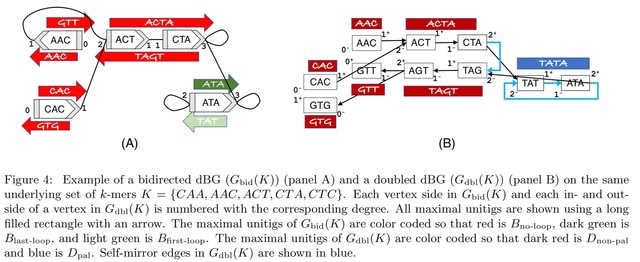
□ Uncovering hidden assembly artifacts: when unitigs are not safe and bidirected graphs are not helpful
>> https://www.biorxiv.org/content/10.1101/2022.01.20.477068v1.full.pdf
Under-assembly issues due to the palindrome artifact are rare in real genomes and, moreover, can be trivially fixed by forcing the unitigs to “push their way through” lonely inverted loops.
A theoretical and empirical study to validate the two hypothesis about common algorithm-driven sources of mis- and under-assemblies. First, despite widespread belief to the contrary, we show that even on error-free data, unitigs do not always appear in the sequenced genome (i.e. they are unsafe).
There is a bijection between maximal unitigs in the doubled and bidirected dBGs, except that palindromic unitigs in the doubled dBG are split in half in the bidirected dBG. Naively using the bidirected graph actually contributes to under-assembly compared to the doubled graph.

□ PSSs: Using syncmers improves long-read mapping
>> https://www.biorxiv.org/content/10.1101/2022.01.10.475696v1.full.pdf
Parameterized Syncmer Schemes provides a theoretical analysis for multiple arbitrary s-minimizer positions. It is possible to retain properties of syncmers such as minimum and most frequent distances b/n selected positions by choosing the correct parameters and downsampling rate.
They incorporates PSSs into the long read mappers minimap2 and Winnowmap2. This syncmer mappers outperformed minimap2 and Winnowmap2 and succeeded in mapping more long reads across a range of different compression values.
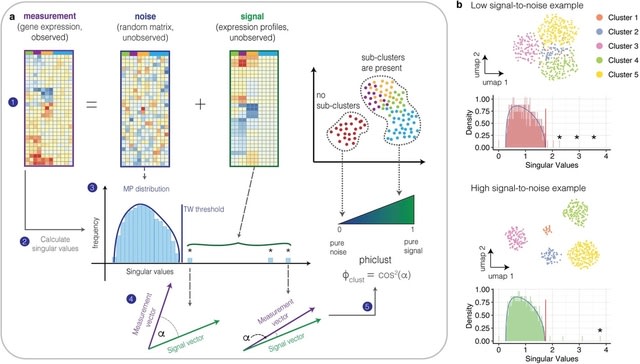
□ Phiclust: a clusterability measure for single-cell transcriptomics reveals phenotypic subpopulations
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02590-x
phiclust (ϕclust), a clusterability measure derived from random matrix theory that can be used to identify cell clusters with non-random substructure, testably leading to the discovery of previously overlooked phenotypes.
Universal properties of the underlying theory make it possible to apply phiclust to arbitrary noise distributions, and the noise can be additive or multiplicative.
If the number of non-zero singular values is small compared to the dimensions of the matrix, low-rank perturbation theory is applicable. This theory allows us to calculate the singular values of the measured gene expression matrix from the singular values of the signal matrix.
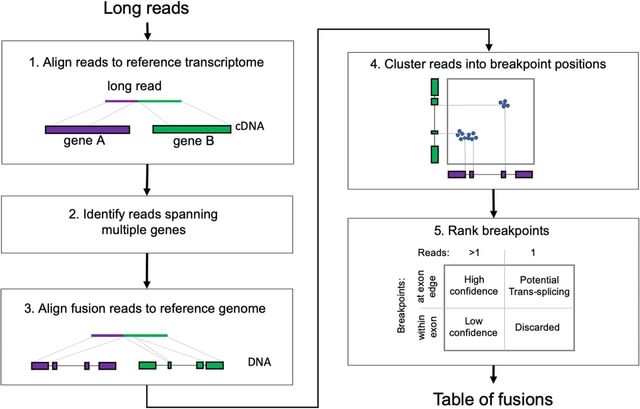
□ JAFFAL: detecting fusion genes with long-read transcriptome sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02588-5
JAFFAL, a new method which is built on the concepts developed in JAFFA and overcomes the high error rate in long-read transcriptome data by using alignment methods and filtering heuristics which are designed to handle noisy long reads.
JAFFAL employs a strategy which anchors transcript breakpoints to exon boundaries. It uses the end position of reference genome alignments to determine fusion breakpoints. JAFFAL is a transcript-centric approach rather than a genome-centric approach like other fusion finders.

□ OLOGRAM-MODL: mining enriched n-wise combinations of genomic features with Monte Carlo and dictionary learning
>> https://academic.oup.com/nargab/article/3/4/lqab114/6478886
OLOGRAM-MODL considers overlaps between n ≥ 2 sets of genomic regions, and computes their statistical mutual enrichment by Monte Carlo fitting of a Negative Binomial distribution, resulting in more resolutive P-values.
OLOGRAM-MODL combines an optional itemset mining algorithm with a statistical model to determine the enrichment of the relevant combinations, asserting whether this combination occurs in the real data across more base pairs that would be expected by chance.
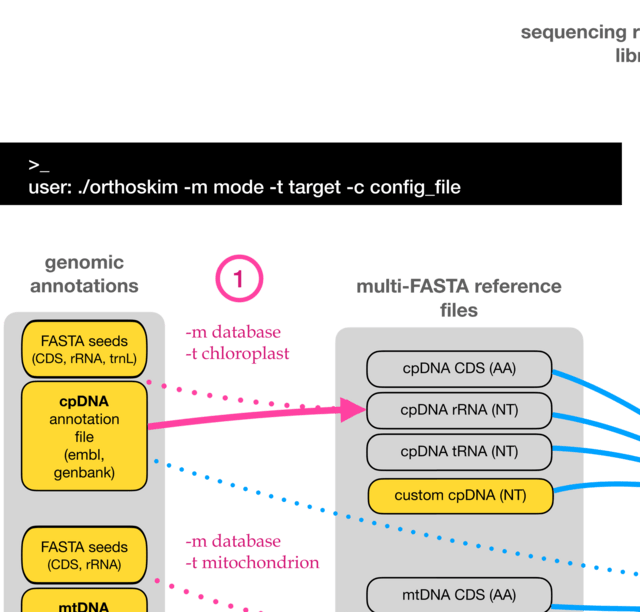
□ ORTHOSKIM: in silico sequence capture from genomic and transcriptomic libraries for phylogenomic and barcoding applications
>> https://onlinelibrary.wiley.com/doi/10.1111/1755-0998.13584
ORTHOSKIM, which performs in silico capture of targeted sequences from genomic and transcriptomic libraries without assembling whole organelle genomes.
ORTHOSKIM proceeds in three steps: 1) global sequence assembly, 2) mapping against reference sequences, and 3) target sequence extraction. ORTHOSKIM recovered with high success rates cpDNA, mtDNA and rDNA sequences.
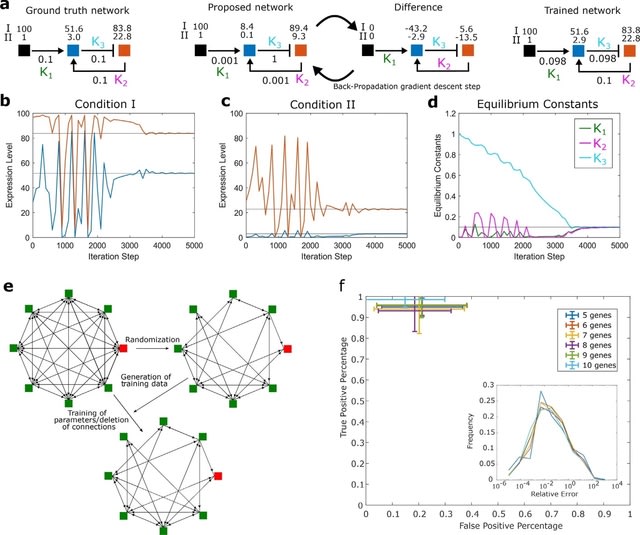
□ CaiNet: Periodic synchronization of isolated network elements facilitates simulating and inferring gene regulatory networks including stochastic molecular kinetics
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04541-6
By considering a deterministic time evolution within each time interval for all elements, this method approaches the solution of the system of deterministic differential equations associated with the GRN.
CaiNet is able to recover the network topology and the network parameters well. CaiNet is able to reproduce noise-induced bi-stability and oscillations in dynamically complex GRNs. This modular approach further allows for a simple consideration of deterministic delays.
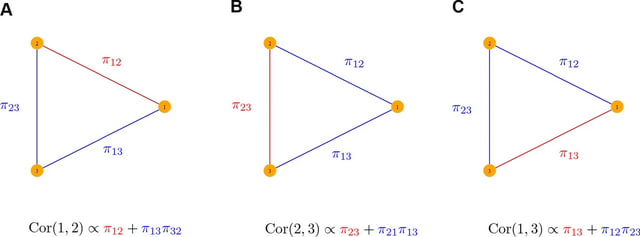
□ PPS: Path-level interpretation of Gaussian graphical models using the pair-path subscore
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04542-5
the pair-path subscore (PPS), a method for interpreting Gaussian graphical models at the level of individual network paths. The scoring is based on the relative importance of such paths in determining the Pearson correlation between their terminal nodes.
The PPS can be used to probe network structure on a finer scale by investigating which paths in a potentially intricate topology contribute most substantially to marginal behavior.
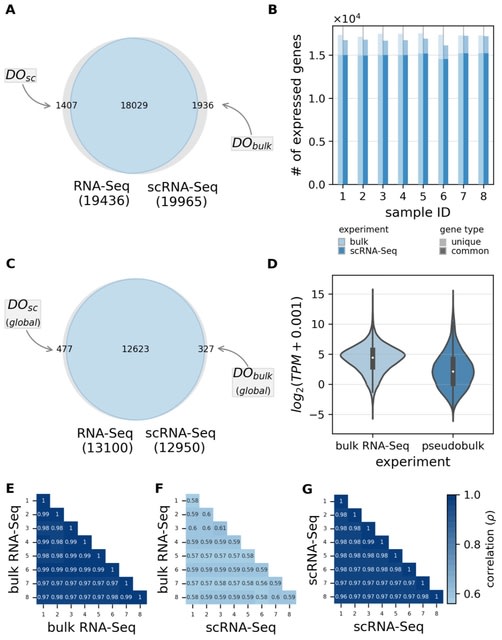
□ FAVSeq: Machine learning-assisted identification of factors contributing to the technical variability between bulk and single-cell RNA-seq experiments
>> https://www.biorxiv.org/content/10.1101/2022.01.06.474932v1.full.pdf
FAVSeq (Factors Affecting Variability in Sequencing data) pipeline analyzes multimodal RNA sequencing data, which allowed to identify factors affecting quantitative difference in gene expression measurements as well as the presence of dropouts.
FAVSeq module supports both non- and parametric imputation strategies, including k-Nearest Neighbors. FAVSeq optimizes hyper-parameters of models through the 5-fold cross-validated (CV) grid-search.
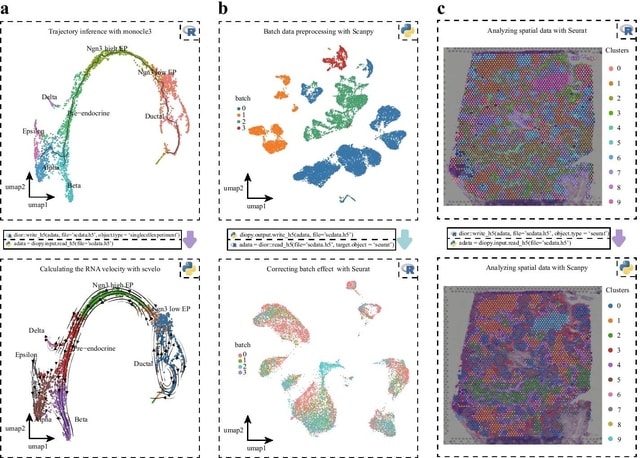
□ scDIOR: single cell RNA-seq data IO software
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04528-3
scDIOR accommodates a variety of data types across programming languages and platforms in an ultrafast way, including single-cell RNA-seq and spatial resolved transcriptomics data, using only a few codes in IDE or command line interface.
scDIOR can perform spatial omics data IO between Seurat and Scanpy. scDIOR creates 8 HDF5 groups to store core single-cell information, including data, layers, obs, var, dimR, graphs, uns and spatial.
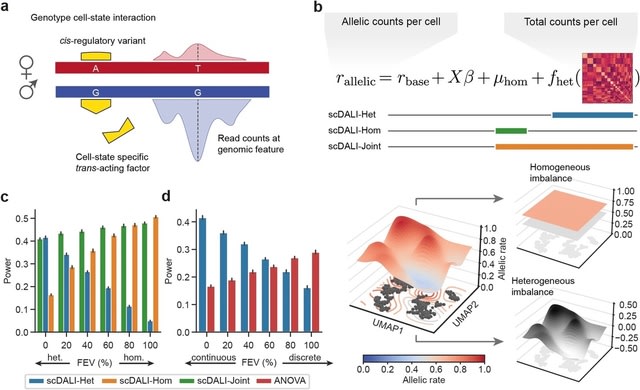
□ scDALI: modeling allelic heterogeneity in single cells reveals context-specific genetic regulation
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02593-8
scDALI, a versatile computational framework that integrates information on cellular states with allelic quantifications of single-cell sequencing data to characterize cell-state-specific genetic effects.
scDALI enables the estimation of allelic imbalance from sparse sequencing data in individual cells, thereby facilitating the visualization and downstream interpretation of allelic regulation.
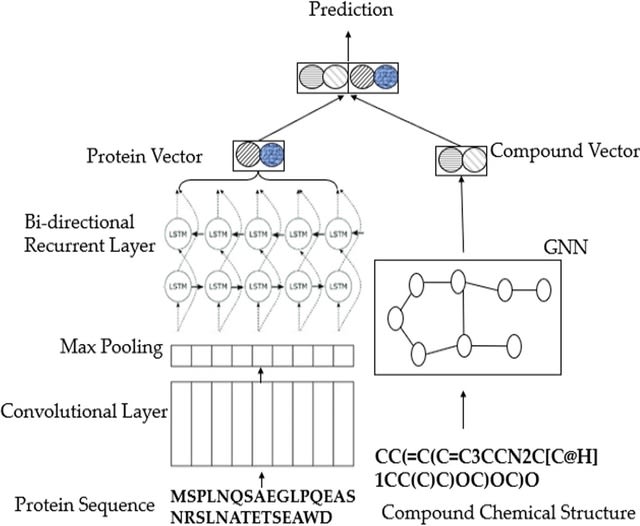
□ GCRNN: graph convolutional recurrent neural network for compound–protein interaction prediction
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04560-x
Graph Convolutional Recurrent Neural Network (GCRNN) uses protein analysis based on a CNN after a max-pooling layer followed by a bidirectional LSTM layer. And Gate Recurrent unit is used for protein sequence vectorization.
GCRNN uses a 3-layer GNN with an r-radius number of 2 to represent molecules as vectors. the CNN takes the original amino acid sequence and passes through a 3-layer structure with 320 convolutional kernels and a window size of 30 with random initiation based on a similar model.
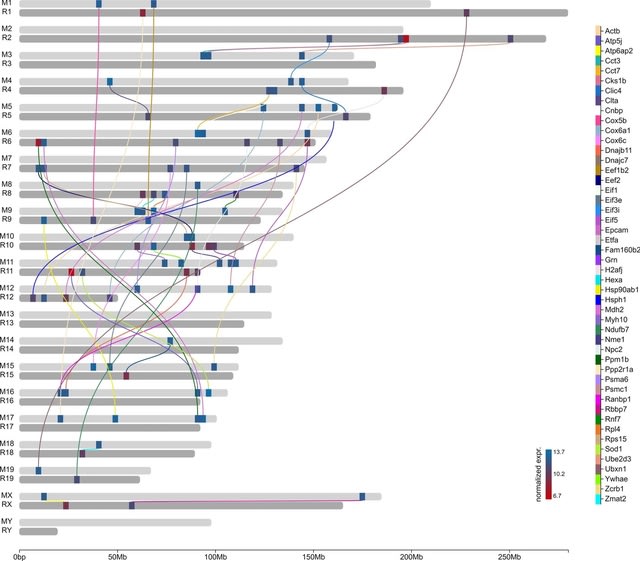
□ ChromoMap: an R package for interactive visualization of multi-omics data and annotation of chromosomes
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04556-z
ChromoMap’s flexibility allows for concurrent visualization of genomic data in each strand of a given chromosome, or of more than one homologous chromosome; allowing the comparison of multi-omic data b/n genotypes or b/n homologous chromosomes of phased diploid/polyploid genomes.
ChromoMap takes tab-delimited files (BED like) or alternatively R objects to specify the genomic co-ordinates of the chromosomes and elements to annotate. ChromoMap renders chromosomes as a continuous composition of windows, to surmount this restriction.
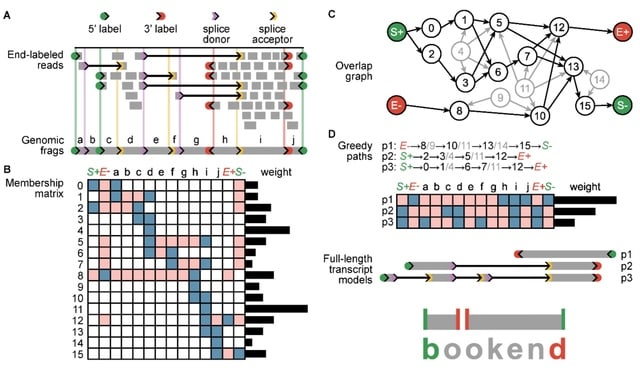
□ Bookend: Precise Transcript Reconstruction with End-Guided Assembly
>> https://www.biorxiv.org/content/10.1101/2022.01.12.476004v1.full.pdf
Bookend uses end information to guide transcript assembly for identifying RNA ends in sequencing data and using the information to assemble transcript isoforms as paths through a network accounting for splice sites, transcription start sites (TSS) and polyadenylation sites (PAS).
Bookend enables the automated annotation of promoter architecture. Bookend takes RNA-seq reads from any method as input and after alignment to a reference genome, reads are stored in a lightweight end-labeled read (ELR) file format that records all RNA boundary features.
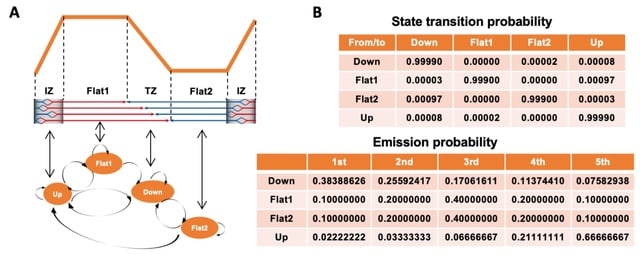
□ OKseqHMM: a genome-wide replication fork directionality analysis toolkit
>> https://www.biorxiv.org/content/10.1101/2022.01.12.476022v1.full.pdf
OKseqHMM directly measures the genome-wide replication fork directionality (RFD) as well as replication initiation and termination from data obtained by Okazaki fragment sequencing (OK-Seq) and related techniques.
OKseqHMM allows accurate detection of replication initiation/termination zones with an HMM algorithm. OKseqHMM can be applied to analyze data obtained by both kinds of techniques, i.e., eSPAN and TrAEL-seq.

□ Telogator: a method for reporting chromosome-specific telomere lengths from long reads
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac005/6505201
While a majority of methods for measuring telomere length will report average lengths across all chromosomes, it is known that aberrations in specific chromosome arms are biomarkers for certain diseases.
Telogator detects chromosome-specific telomere length in simulated data across a range of read lengths and error rates. And investigate common subtelomere rearrangements and identify the minimum read length required to anchor telomere/subtelomere boundaries.

□ c-TSNE: Explainable t-SNE for single-cell RNA-seq data analysis
>> https://www.biorxiv.org/content/10.1101/2022.01.12.476084v1.full.pdf
c-TSNE (cell-driven t-SNE), an explainable t-SNE that demonstrates robustness to dropout and noise in dimension reduction and clustering. It provides a novel and practical way to investigate the interpretability of t-SNE in scRNA-seq data analysis.
c-TSNE uses appropriate and explainable distance metrics incl. Yule, L-Chebyshev, and fractional distance metrics. The cell-driven distance metrics make more relevant samples mapped as the closest neighbors to each other in the low-dimensional embedding space.
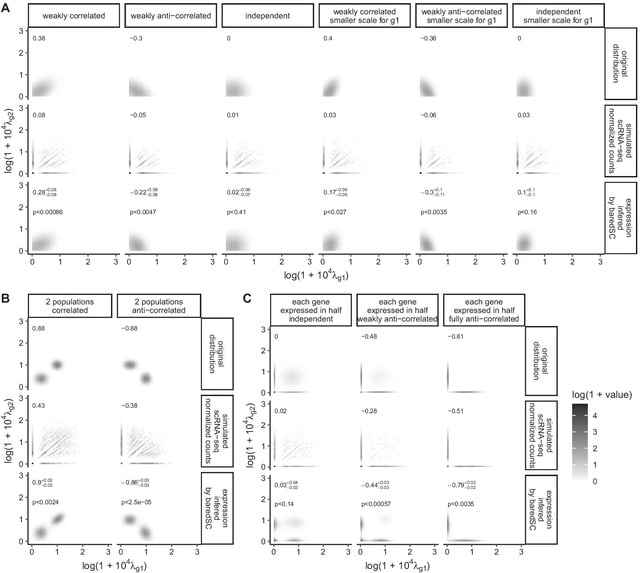
□ baredSC: Bayesian approach to retrieve expression distribution of single-cell data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04507-8
baredSC infers the intrinsic expression distribution using a Gaussian mixture model. baredSC can be used to obtain the distribution in one dimension for individual genes and in two dimensions for pairs of genes, in particular to estimate the correlation in the two genes.
baredSC allows to retrieve precisely multi-modal expression distribution even when they are not distinguishable in the input data due to sampling noise. And is able to uncover the expression distribution used to simulate the data, even in multi-modal cases with very sparse data.

□ A semi-supervised Bayesian mixture modelling approach for joint batch correction and classification
>> https://www.biorxiv.org/content/10.1101/2022.01.14.476352v1.full.pdf
This model allows observations to be probabilistically assigned to classes in a way that incorporates uncertainty arising from batch effects.
The MVN mixture model exhibited good behaviour, except when misspecified as in the MVT generated data. the MVT mixture model’s estimate tended to be either centred on the true value.

□ LYRUS: a machine learning model for predicting the pathogenicity of missense variants
>> https://academic.oup.com/bioinformaticsadvances/article-abstract/2/1/vbab045/6483096
LYRUS, a machine learning method that uses an XGBoost classifier to predict the pathogenicity of SAVs. LYRUS incorporates five sequence-based, six structure-based and four dynamics-based features.
LYRUS includes a newly proposed sequence co-evolution feature called the variation number. Variation numbers employed in the model are scaled using min. to max. normalization for each amino acid sequence.
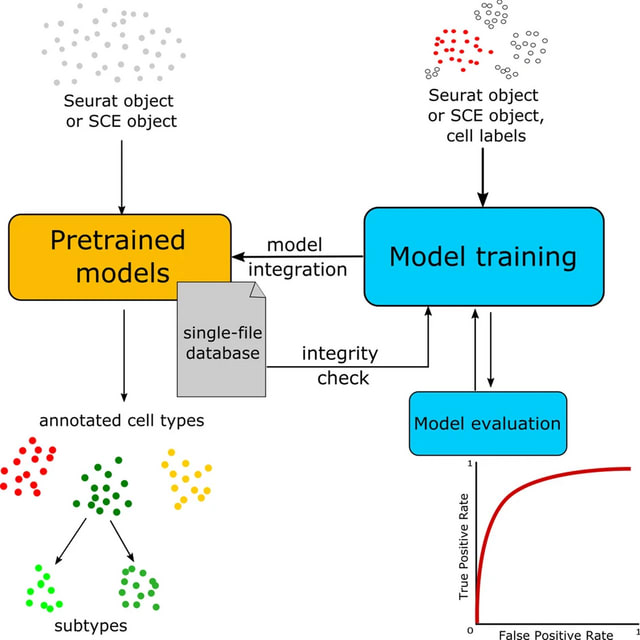
□ scAnnotatR: framework to accurately classify cell types in single-cell RNA-sequencing data
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04574-5
scAnnotatR is a novel R package that provides a complete framework to classify cells in scRNA-seq datasets using pre-trained classifiers. It supports both Seurat and Bioconductor’s SingleCellExperiment and is thereby compatible w/ the vast majority of R-based analysis workflows.
scAnnotatR uses hierarchically organised SVMs to distinguish a specific cell type versus all others. It shows comparable or even superior accuracy, sensitivity and specificity compared to existing tools while being able to not-classify unknown cell types.
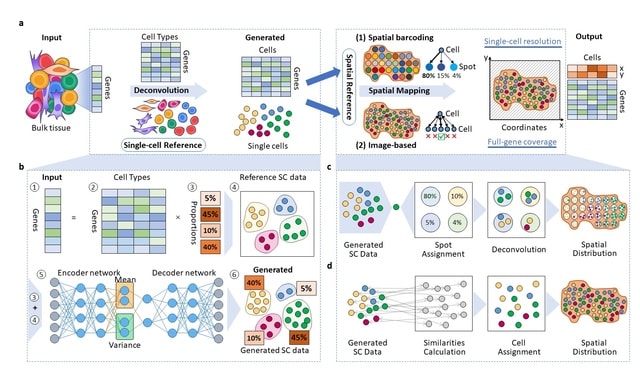
□ Bulk2Space: Spatially resolved single-cell deconvolution of bulk RNA-seq
>> https://www.biorxiv.org/content/10.1101/2022.01.15.476472v1.full.pdf
Bulk2Space, a spatial deconvolution method based on deep learning frameworks, which converts bulk transcriptomes into spatially resolved single-cell expression profiles using existing high-quality scRNA-seq data and spatial transcriptomics as references.
Bulk2Space first generates single-cell transcriptomic data within the clustering space to find a set of cells whose aggregated data are close to the bulk data. Next, the generated single cells were allocated to optimal spatial locations using a spatial transcriptome reference.
□ A novel gene functional similarity calculation model by utilizing the specificity of terms and relationships in gene ontology
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04557-6
The proposed method mainly contains three steps. Firstly, a novel computing model is put forward to compute the IC of terms. This model has the ability to exploit the specific structural information of GO terms.
Secondly, the IC of term sets are computed by capturing the genetic structure between the terms contained in the set.
They measure the gene functional similarity according to the IC overlap ratio of the corresponding annotated genes sets. The proposed method accurately measures the IC of not only GO terms but also the annotated term sets by leveraging the specificity of edges in the GO graph.
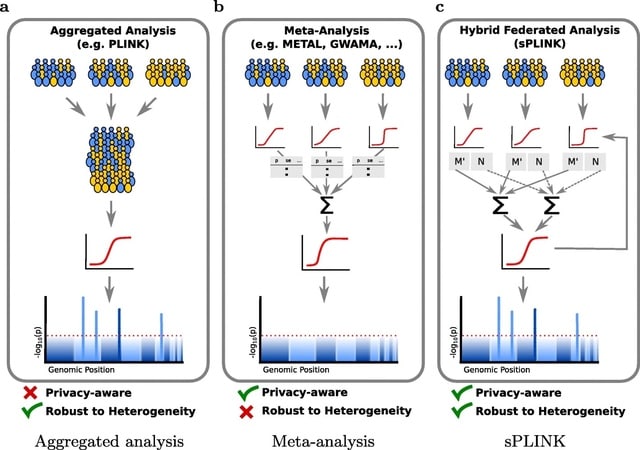
□ sPLINK: a hybrid federated tool as a robust alternative to meta-analysis in genome-wide association studies
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02562-1
sPLINK, a hybrid federated and user-friendly tool, which performs privacy-aware GWAS on distributed datasets while preserving the accuracy of the results.
sPLINK is robust against heterogeneous distributions of data across cohorts while meta-analysis considerably loses accuracy in such scenarios. sPLINK achieves practical runtime and acceptable network usage for chi-square and linear/logistic regression tests.
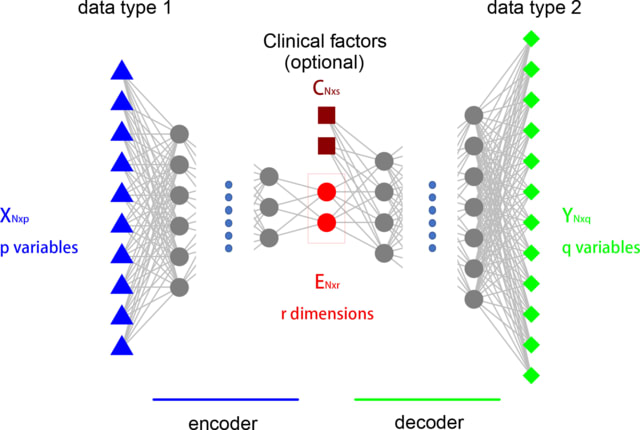
□ AIME: Autoencoder-based integrative multi-omics data embedding that allows for confounder adjustments
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009826
AIME can detect nonlinear associations between the data matrices. It finds data embedding from the input data matrix that best preserves its relation with the output data matrix.
AIME can be seen as a nonlinear equivalent to CCA, with the added capability to adjust for confounder variables. AIME is even more effective than traditional linear methods such as CCA, PLS, jSVD, iCluster2 and MOFA2 in extracting linear relationships.

□ LmTag: functional-enrichment and imputation-aware tag SNP selection for population-specific genotyping arrays
>> https://www.biorxiv.org/content/10.1101/2022.01.28.478108v1.full.pdf
LmTag, a novel method for tag SNP selection that not only improves imputation performance but also prioritizes highly functional SNP markers.
LmTag uses a robust statistical modeling to systematically integrate LD information, minor allele frequency (MAF), and physical distance of SNPs into the imputation accuracy score to improve tagging efficiency.
LmTag adapts the beam search framework to prioritize both variant imputation scores and functional scores to solve the tag SNP selection problem. LmTag improves both imputation performance and prioritization of functional variants.
Tagging efficiency of tag SNP sets selected by LmTag are sustainability higher than existing genotyping arrays, indicating the potential improvements for future genotyping platforms.
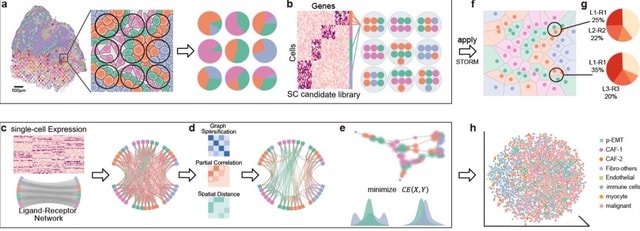
□ STORM: spectral sparsification helps restore the spatial structure at single-cell resolution
>> https://www.biorxiv.org/content/10.1101/2022.01.25.477389v1.full.pdf
STORM reconstructs the single-cell resolution quasi-structure from the spatial transcriptome with diminished pseudo affinities.
STORM first curates the representative single-cell profiles for each spatial spot from a candidate library, then reduces the pseudo affinities in the intercellular affinity matrix by partial correlation, spectral graph sparsification, and spatial coordinates refinement.
STORM embeds the estimated interactions into a low-dimensional space with the cross-entropy objective to restore the intercellular quasi-structures, which facilitates the discovery of dominant ligand-receptor pairs between neighboring cells at single-cell resolution.
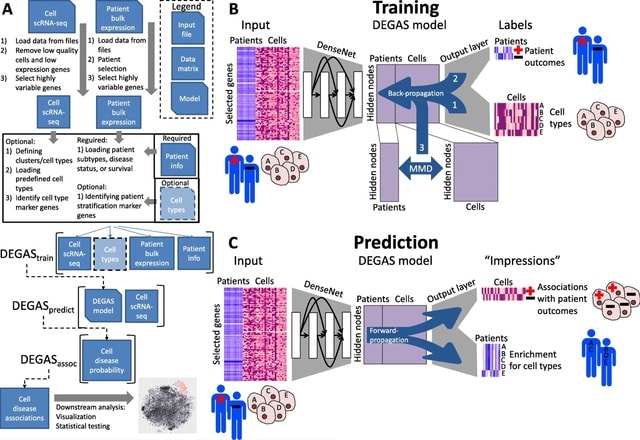
□ Diagnostic Evidence GAuge of Single cells (DEGAS): a flexible deep transfer learning framework for prioritizing cells in relation to disease
>> https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-022-01012-2
DEGAS, the deep transfer learning framework to integrate scRNA-seq and patient-level transcriptomic data in order to infer the transferrable “impressions” between patient characteristics in single cells and cellular characteristics in patients.
DEGAS models are trained using both single-cell and patient disease attributes using a multitask learning neural network that learns latent representation reducing the differences between patients and single cells at the final hidden layer using Maximum Mean Discrepancy.
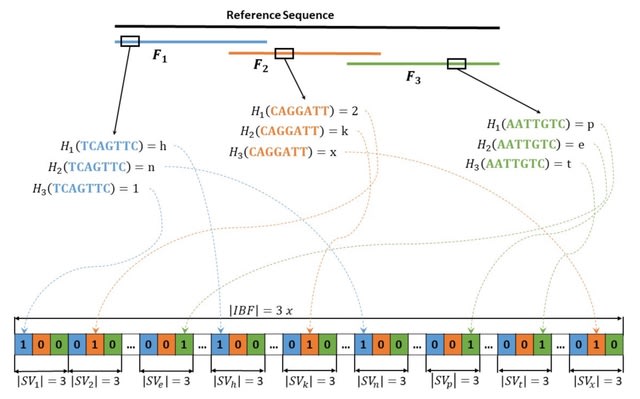
□ ReadBouncer: Precise and Scalable Adaptive Sampling for Nanopore Sequencing
>> https://www.biorxiv.org/content/10.1101/2022.02.01.478636v1.full.pdf
Read-Bouncer, a new approach for nanopore adaptive sam- pling that combines fast CPU and GPU base calling with read classification based on Interleaved Bloom Filters (IBF).
ReadBouncer uses Oxford Nanopore's Read Until functionality to unblock reads that match to a given reference sequence database. Signals are basecalled in real-time with Guppy or DeepNano-blitz.









