









(made with DALL-E 2)
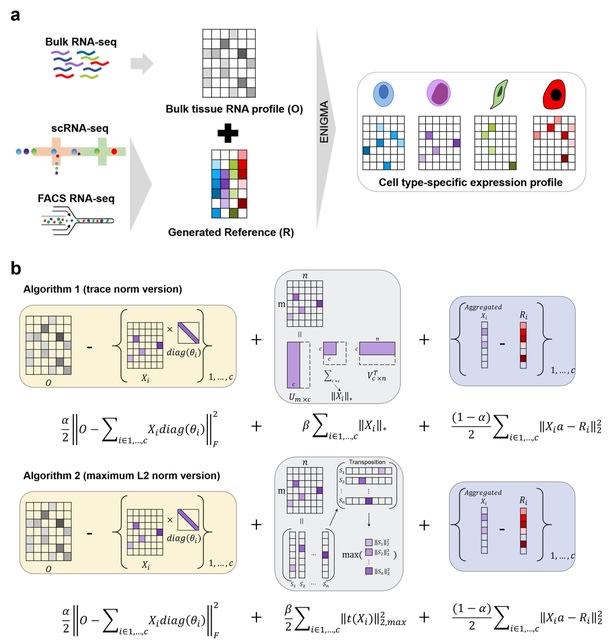
□ ENIGMA: Approximate estimation of cell-type resolution transcriptome in bulk tissue through matrix completion
>> https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbad273/7234627
ENIGMA (Deconvolution based on Regularized Matrix Completion), a method that addresses this limitation through accurately deconvoluting bulk tissue RNA-seq data into a readout with cell-type resolution by leveraging information from scRNA-seq data.
ENIGMA employs a matrix completion strategy to minimizes the distance between the mixture transcriptome obtained with bulk sequencing and a weighted combination of cell-type-specific expression. ENIGMA reconstructs the latent continuous structure of CSE into a pseudo-trajectory.
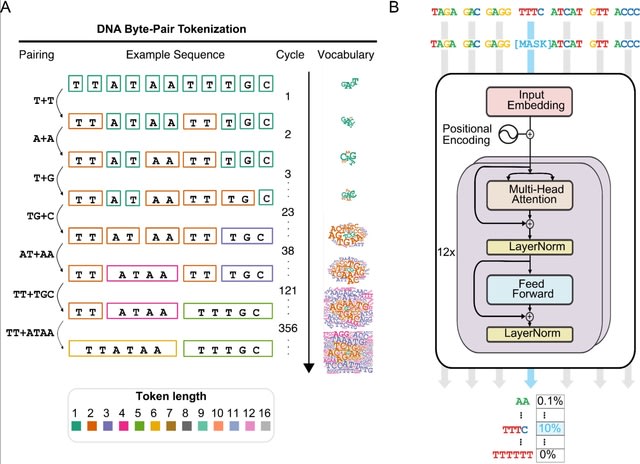
□ GROVER: The human genome’s vocabulary as proposed by the DNA language model
>> https://www.biorxiv.org/content/10.1101/2023.07.19.549677v1
GROVER ("Genome Rules Obtained Via Extracted Representations") to select the optimal vocabulary with a custom fine-tuning task of next-k-mer prediction. GROVER has learned these structures purely from the contextual relationships of tokens.
GROVER extracts the information content of the genome, its language structures via token embeddings or through extracting attention from the foundation model. Self-similarity was assessed as the cosine similarity of different embeddings separately for all 12 transformer layers.

□ biomolecular neuron: Simple and rewireable biomolecular building blocks for DNA machine-learning algorithms
>> https://www.biorxiv.org/content/10.1101/2023.07.20.549967v1
biomolecular neuron, a polymerase-actuated DNA computing unit which serve as rewireable building blocks for neural network algorithms. biomolecular neuron generates DNA computing units of longer lengths than is feasible via chemical synthesis.
This scheme combines enzymatic synthesis to encode a greater number of i/o connections on a single DNA strand, solid-phase immobilization to spatially segregate DNA computing units into network layers, and universal addressing to enable the assembly of different circuits.
biomolecular neuron generates computing units from fewer DNA sequences, and built-in modularity through circuit rewiring. a surface-based DNA computing approach has a unique feature: computation at each layer is synchronized to the timing of fluid transfer.
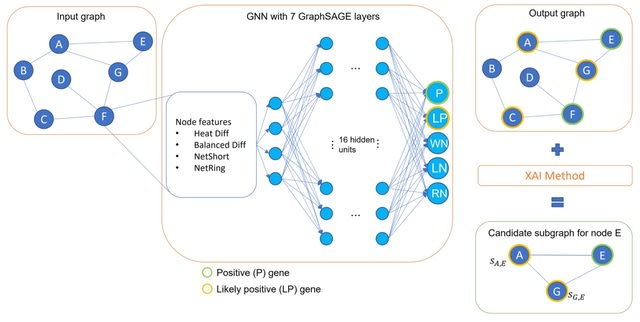
□ XGDAG: eXplainable Gene–Disease Associations via Graph Neural Networks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad482/7235567
XGDAG is the first method to use an XAI-based solution in the context of positive-unlabeled learning for disease gene prioritization with GNNs. A graph based on a PPI network and enriched with GDA information and node features is fed into a graph neural network.
XGDAG exploits XAI methods to draw the final ranking of candidate genes. This is a novelty that presents XAI not only as a tool that opens the black box of deep neural networks but also as an analysis component directly incorporated into the GDA discovery pipeline.
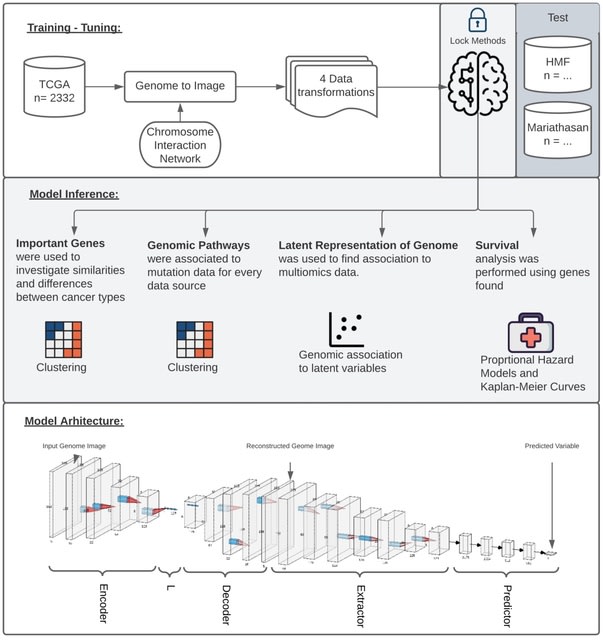
□ GENIUS: GEnome traNsformatIon and spatial representation of mUltiomicS data
>> https://www.biorxiv.org/content/10.1101/2023.02.09.525144v3
Integrated Gradients evaluates the trained model relative to I/O label, resulting in attribution scores for each input w/ respect to the output label. Integrated Gradients represent the integral of gradients with respect to inputs along the path from a given baseline.
GENIUS (GEnome traNsformatIon and spatial representation of mUltiomicS data) can transform multi-omics data into images with genes displayed as spatially connected pixels and successfully extract relevant information with respect to the desired output.
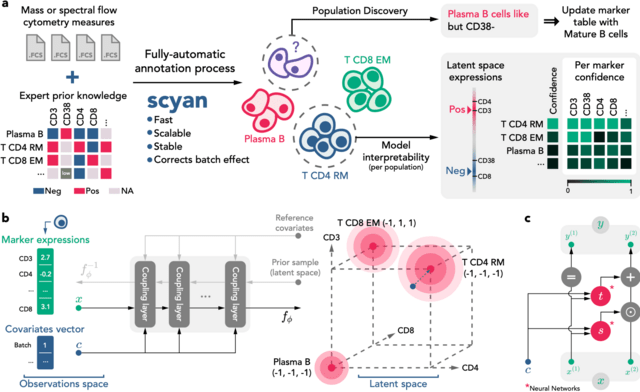
□ scyan: Biology-driven deep generative model for cell-type annotation in cytometry
>> https://mics-lab.github.io/scyan/
Scyan (Single-cell Cytometry Annotation Network) is a Bayesian probabilistic model composed of a deep invertible neural network called a normalizing flow (the function ). It maps a latent distribution of cell expressions into the empirical distribution of cell expressions.
This cell distribution is a mixture of gaussian-like distributions representing the sum of a cell-specific and a population-specific term. Also, interpretability and batch effect correction are based on the model latent space.
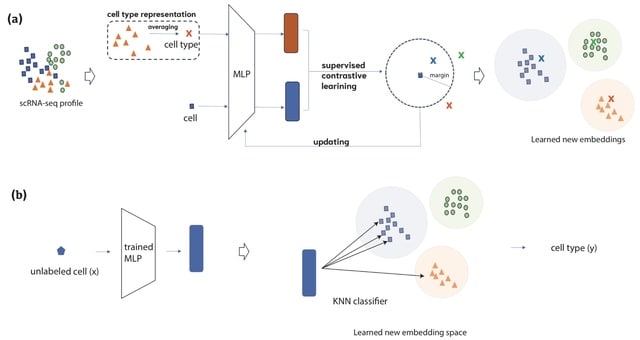
□ SCLSC: Predicting cell types with supervised contrastive learning on cells and their types
>> https://www.biorxiv.org/content/10.1101/2023.08.08.552379v1
SCLSC (Supervised Contrastive Learning for Single Cell) leverages supervised contrastive learning, which utilizes label information from the training data to provide explicit guidance on the similarity or dissimilarity between samples during the learning process.
SCLSC has two key parameters: the dimension of the input and the dimension of the output of the encoder. In case of input dimension, SCLSC has the capability to process input from all genes.
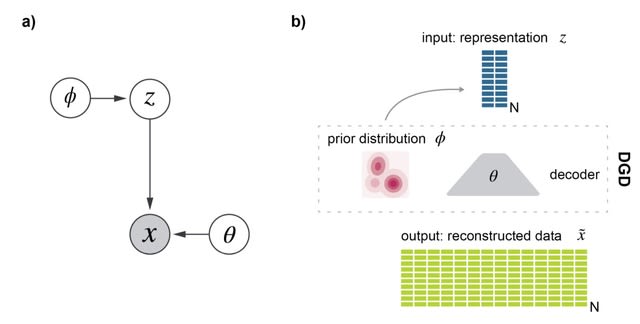
□ scDGD: The Deep Generative Decoder: MAP estimation of representations improves modeling of single-cell RNA data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad497/7241685
scDGD is an application of the encoder-less generative model, the Deep Generative Decoder (DGD), a simple generative model that computes model parameters and representations directly via maximum a posteriori (MAP) estimation.
The DGD handles complex parameterized latent distributions naturally unlike VAEs which typically use a fixed Gaussian distribution, because of the complexity of adding other types.
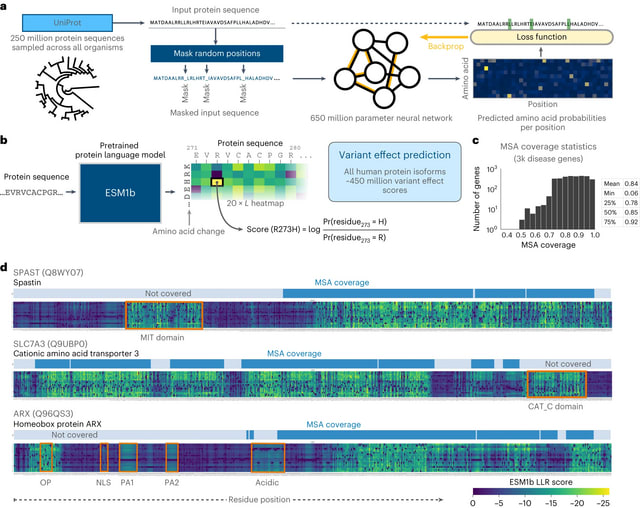
□ Genome-wide prediction of disease variant effects with a deep protein language model
>> https://www.nature.com/articles/s41588-023-01465-0
ESM1b, a 650-million-parameter protein language model trained on 250 million protein sequences. The model was trained via the masked language modeling task, where random residues are masked from input sequences and the model has to predict the correct amino acid at each position.
ESM1b computes the LLR scores for all possible missense mutations in a protein through a single pass.
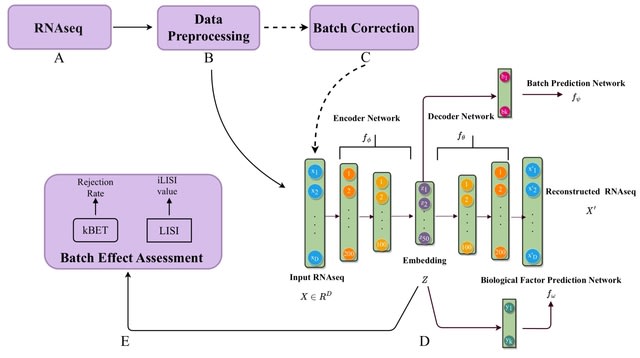
□ BEENE: Deep Learning based Nonlinear Embedding Improves Batch Effect Estimation https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad479/7240486
BEENE uses an autoencoder model to learn the nonlinear embeddings of RNA-seq expression data. The nonlinear embedding learned by the autoencoder is used by both batch and biological variable learner modules.
The autoencoder and these two learning networks are trained in tandem to guide the embedding in such a way that biological heterogeneity in the data as well as variability across batches are preserved.
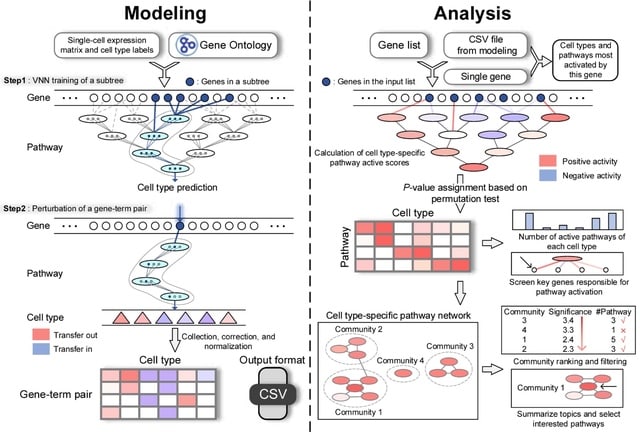
□ CellGO: A novel deep learning-based framework and webserver for cell type-specific gene function interpretation
>> https://www.biorxiv.org/content/10.1101/2023.08.02.551654v1
CellO, a VNN-based tool for cell type-specific pathway analysis. CellGO integrates the single-cell RNA expression data and the VNN model that emulates the hierarchy of GO terms to capture cell type-specific signatures, intra-pathway gene connections, and inter-pathway crosstalk.
CellO can construct the network of cell type-specific active pathways and report top communities enriched with active pathways, by incorporating the random walk with restart algorithm and the community partition algorithm.
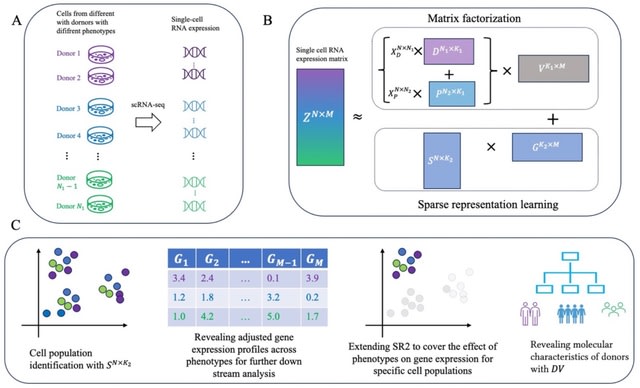
□ SR2: Sparse Representation Learning for Scalable Single-cell RNA Sequencing Data Analysis
>> https://www.biorxiv.org/content/10.1101/2023.07.31.551228v1
SR2 is based on an ensemble of matrix factorization and sparse representation learning. It decomposes variation from multiple biological conditions and cellular variation across bio-samples into shared low-rank latent spaces.
SR2 employs sparse regularization on embedding of cells to facilitate cell population discovery and norm constraint on each component of gene representations to ensure equal scale.
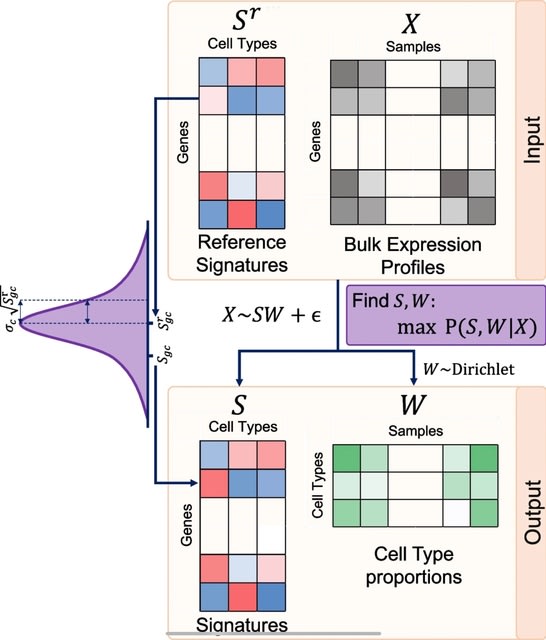
□ BEDwARS: a robust Bayesian approach to bulk gene expression deconvolution with noisy reference signatures
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03007-7
BEDwARS (Bayesian Expression Deconvolution with Approximate Reference Signatures), which tackles the problem of signature mismatch from a complementary angle.
BEDwARS incorporates the possibility of reference signature mismatch directly into the statistical model used for deconvolution, using the reference to estimate the true cell type signatures underlying the given bulk profiles while simultaneously learning cell type proportions.
BEDwARS assumes that each bulk expression profile is a weighted mixture of cell type-specific profiles (“true signatures”) that are unknown but not very different from given reference signatures.
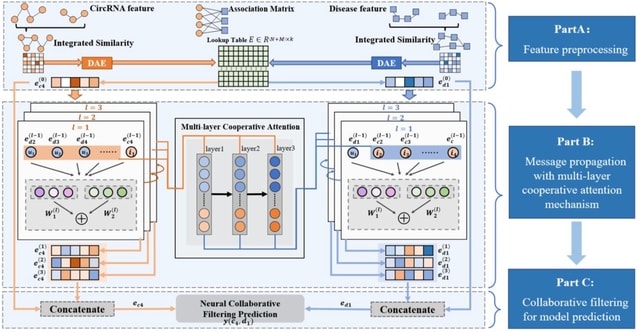
□ MLNGCF: circRNA-disease associations prediction with multi-layer attention neural graph based collaborative filtering
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad499/7240485
MLNGCF first enhances multiple biological information with autoencoder as the initial features of circRNAs and diseases. A multi-layer cooperative attention-based message propagation is performed on the central network to obtain the high-order features of circRNAs and diseases.
An interaction function of collaborative filtering is introduced to integrate both matrix factorization and multilayer perceptron and score circRNAs-disease associations.
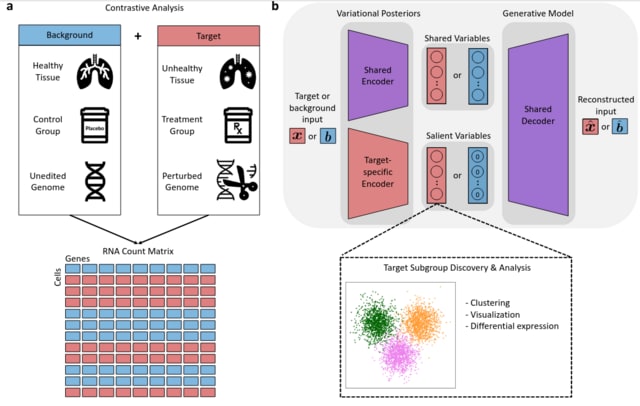
□ contrastiveVI: Isolating salient variations of interest in single-cell data
>> https://www.nature.com/articles/s41592-023-01955-3
contrastiveVI (contrastive Variational Inference), a framework for deconvolving variations in treatment–control single-cell RNA sequencing (scRNA-seq) datasets into shared and treatment-specific latent variables.
contrastiveVI is a generative model designed to isolate factors of variation specific to a group of "target" cells (e.g. from specimens with a given disease) from those shared with a group of "background".
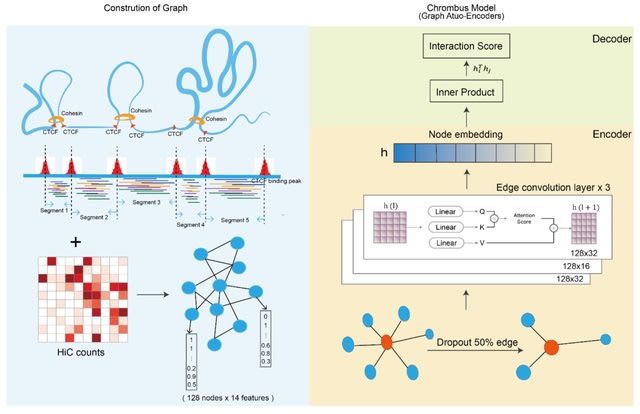
□ Chrombus-XMBD: A Graph Generative Model Predicting 3D-Genome, ab initio from Chromatin Features
>> https://www.biorxiv.org/content/10.1101/2023.08.02.551072v1
Chrombus-XMBD, a graph generative model capable of predicting chromatin interactions. Chrombus employes dynamic edge convolution with QKV attention setup, which maps the relevant chromatin features to a learnable embedding space thereby generate genome-wide 3D-contactmap.
Chrombus is adopted from Graph Auto-Encoder architecture. Each graph consists 128 vertices, and each vertex represents a chromatin segment derived from CTCF binding peaks. The node (vertex) attributes consist 14-dimensional chromatin features.
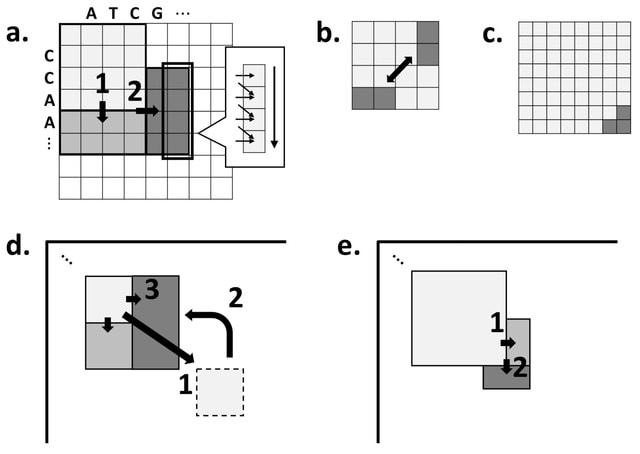
□ Block Aligner: an adaptive SIMD-accelerated aligner for sequences and position-specific scoring matrices
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad487/7236499
Block Aligner, a new SIMD-accelerated algorithm for aligning nucleotide and protein sequences against other sequences or position-specific scoring matrices. They introduce a new paradigm that uses blocks in the dynamic programming matrix that greedily shift, grow, and shrink.
Block Aligner relies on the COMPUTE_RECT function to efficiently compute the scores for certain rectangular regions of the DP matrix. Block Aligner generally tries to center the maximum scores (likely optimal alignment path) within the computed regions.
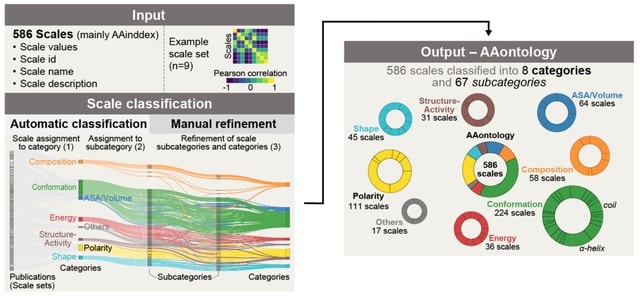
□ AAontology: An ontology of amino acid scales for interpretable machine learning
>> https://www.biorxiv.org/content/10.1101/2023.08.03.551768v1
AAontology-a two-level classification for 586 amino acid scales (mainly from AAindex) together with an in-depth analysis of their relations-using bag-of-word-based classification, clustering, and manual refinement over multiple iterations.
AAontology organizes amino acid property scales into 8 categories and 67 subcategories based on their numerical similarity and physicochemical meaning.
The Energy category comprises around 40 scales organized into 9 specific subcategories, each highlighting different energetic aspects of amino acids including free energy determining conformational stability.
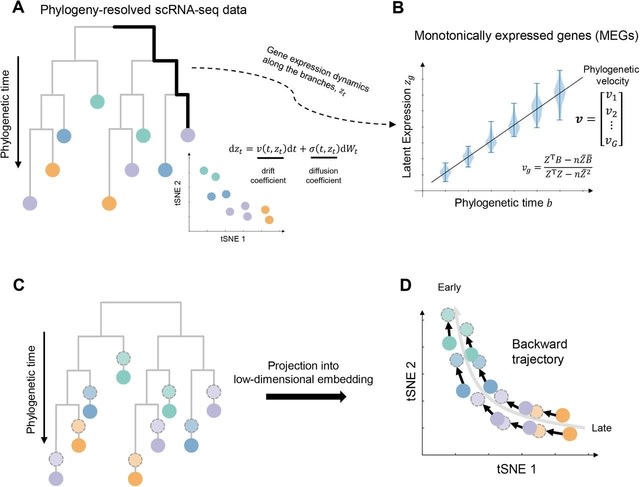
□ PhyloVelo enhances transcriptomic velocity field mapping using monotonically expressed genes
>> https://www.nature.com/articles/s41587-023-01887-5
PhyloVelo, a computational framework that estimates the velocity of transcriptomic dynamics by using monotonically expressed genes (MEGs) or genes with expression patterns that either increase or decrease, but do not cycle, through phylogenetic time.
PhyloVelo identifies MEGs and reconstructs a transcriptomic velocity field. A diffusion process is used to model the dynamics of latent gene expression. This enables the estimation of phylogenetic velocity, which corresponds to the drift coefficients of MEGs in the diffusion process.
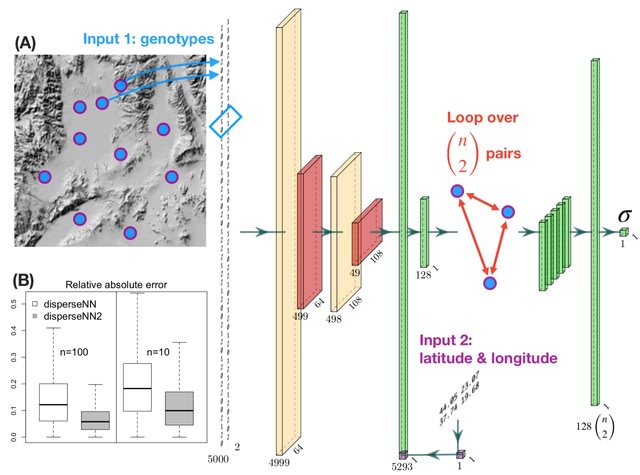
□ disperseNN2: a neural network for estimating dispersal distance from georeferenced polymorphism data
>> https://www.biorxiv.org/content/10.1101/2023.07.30.551115v1
The disperseNN2 program uses a deep neural network trained on simulated data to infer the mean, per-generation parent-offspring distance. It aims to infer σ, the root-mean-square displacement along a given axis between a randomly chosen child and one of their parents chosen at random.
disperseNN2 is designed for SNP data obtained from RADseq or whole genome sequencing, with either short-range or full linkage information. disperseNN2 uses a pairwise convolutional network that performs feature extraction on pairs of individuals at a time.
“The extractor" extracts pertinent information from pairs of genotypes, and merges the extracted features from all combinatorial pairs into a summary table for downstream processing.
This strategy allows us to convey spatial information to the network which is accomplished by attaching the geographic distance between each sample-pair directly to the genotype summaries from the corresponding pair.
The first input to disperseNN2 is a genotype matrix consisting of minor allele counts (Os, Is, and 2s) for m SNPs from n individuals. However, rather than show the full genotype matrix to the network, it loops through all pairs of individuals and sub-set the genotypes of each pair.

□ PACS: Model-based compound hypothesis testing for snATAC-seq data
>> https://www.biorxiv.org/content/10.1101/2023.07.30.551108v1
PACS (Probability model of Accessible Chromatin of Single cells), a zero-adjusted statistical model that can allow complex hypothesis testing of factors that affect accessibility while accounting for sparse and incomplete data.
PACS could detect both linear and quadratic signals, and its power is dependent on the "effect sizes" defined as the log fold change of accessibility between the highest and lowest accessibility.
PACS resolves the issue of sequencing coverage variability in scATAC-seq data by combining a probability model of the underlying group-level accessibility with an independent cell-level capturing probability.
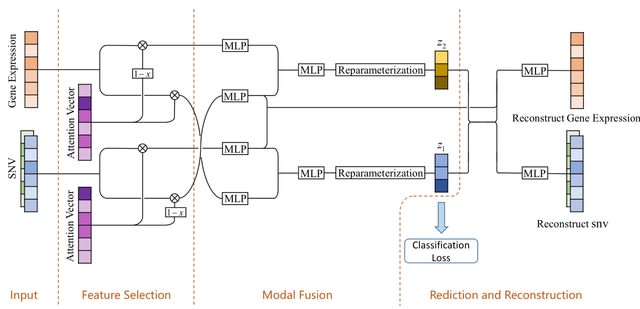
□ ISMI-VAE: A Deep Learning Model for Classifying Disease Cells Using Gene Expression and SNV Data
>> https://www.biorxiv.org/content/10.1101/2023.07.28.550985v1
ISMI-VAE leverages latent variable models that utilize the characteristics of SNV and gene expression data to overcome high noise levels, and uses deep learning techniques to integrate multimodal information, map them to a low-dimensional space, and classify disease cells.
ISMI-VAE combines attention mechanism and variational autoencoder. It proposes an attention module that uses the weights of the attention vector to reflect the importance of gene features as a way to determine genes or SNVs that are highly associated with disease.
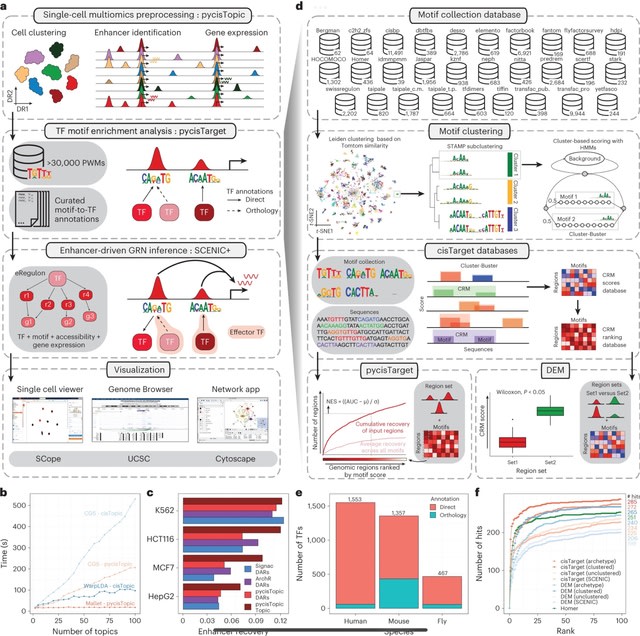
□ SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks
>> https://www.nature.com/articles/s41592-023-01938-4
SCENIC+, a computational framework that combines single-cell chromatin accessibility and gene expression data with motif discovery to infer enhancer-driven GRNs.
SCENIC+ integrates region accessibility, TF and target gene expression and cistromes to infer eGRNs, in which TFs are linked to their target regions and these to their target genes.
SCENIC+ next uses GRNBoost2 to quantify the importance of both TFs and enhancer candidates for target genes and it infers the direction of regulation (activating/repressing) using linear correlation.
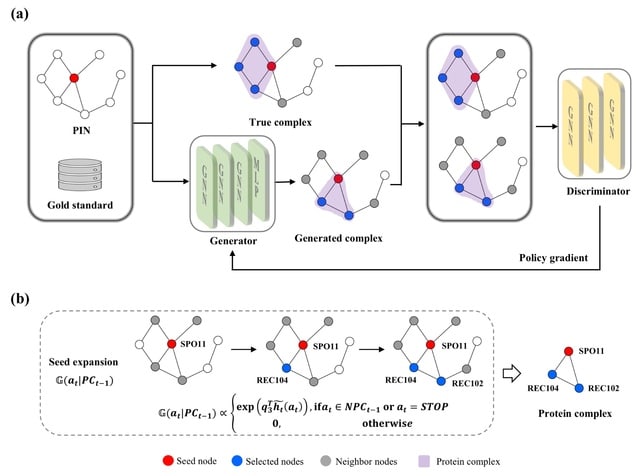
□ PCGAN: A Generative Approach for Protein Complex Identification from Protein Interaction Networks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad473/7235566
PCGAN (Protein Complexes by Generative Adversarial Networks) learns the characteristics of complexes, and generates new complexes. PCGAN trains a generator for generating protein complexes, and a discriminator for distinguishing the generated protein complexes from real ones.
The input data of PCGAN includes a PIN and a gold standard dataset. The competition learning between the generator and the discriminator promotes the two models to improve their capabilities until the generated complexes are indistinguishable from the real ones.
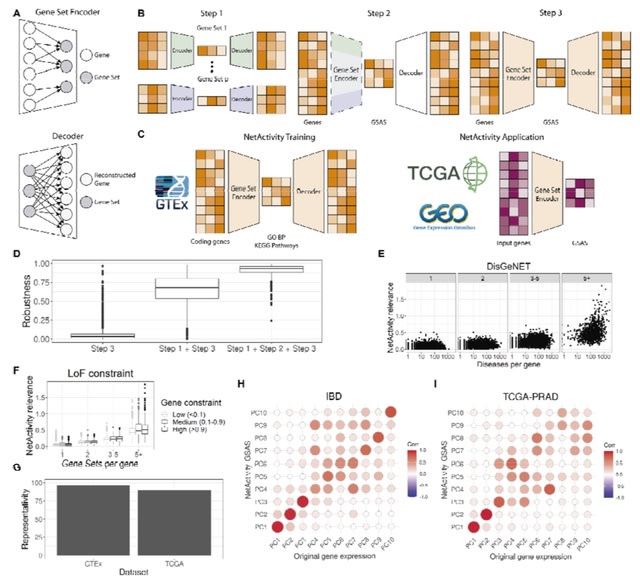
□ NetActivity enhances transcriptional signals by combining gene expression into robust gene set activity scores through interpretable autoencoders
>> https://www.biorxiv.org/content/10.1101/2023.07.31.551238v1
NetActivity, a computational framework to define highly representative and interpretable gene set activity scores (GSAS) based on shallow sparsely-connected autoencoders. NetActivity model was trained w/ 1,518 GO biological processes terms and KEGG pathways and all GTEx samples.
NetActivity generates GSAS robust to the initialization parameters and representative of the original transcriptome, and assigned higher importance to more biologically relevant genes. NetActivity returns GSAS w/ a more consistent definition and higher interpretability than GSVA.
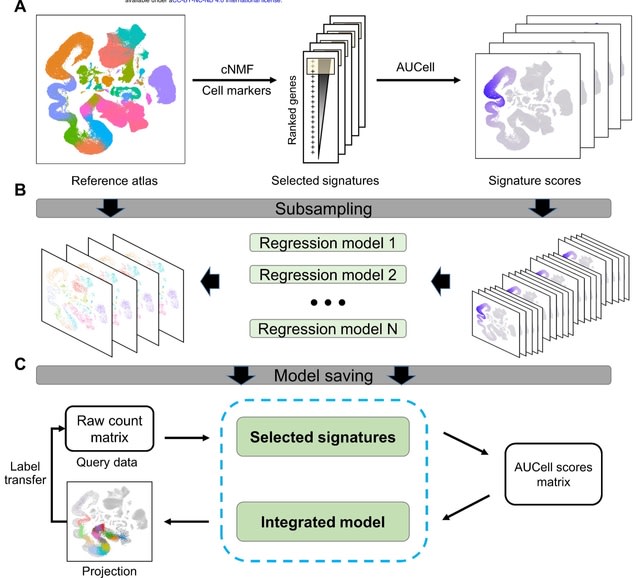
□ ProjectSVR: Mapping single-cell RNA-seq data to reference atlases by supported vector regression
>> https://www.biorxiv.org/content/10.1101/2023.07.31.551202v1
ProjectSVR, a machine learning-based algorithm for mapping the query cells onto well-constructed reference embeddings using Supported Vector Regression.
ProjectS VR follows a two-step process for reference mapping: (1) Fitting a collection of SR model ensembles to learn embeddings from feature scores of the reference atlas; (2) Projecting the query cells onto the consistent embeddings of the reference via trained SR models.

□ BigSeqKit: a parallel Big Data toolkit to process FASTA and FASTQ files at scale
>> https://academic.oup.com/gigascience/article/doi/10.1093/gigascience/giad062/7233988
BigSeqKit, a parallel toolkit to manipulate FASTA and FASTQ files at scale with speed and scalability at its core.
BigSeqKit takes advantage of IgnisHPC, a computing engine that unifies the development, combination, and execution of high-performance computing (HPC) and Big Data parallel tasks using different languages and programming models.
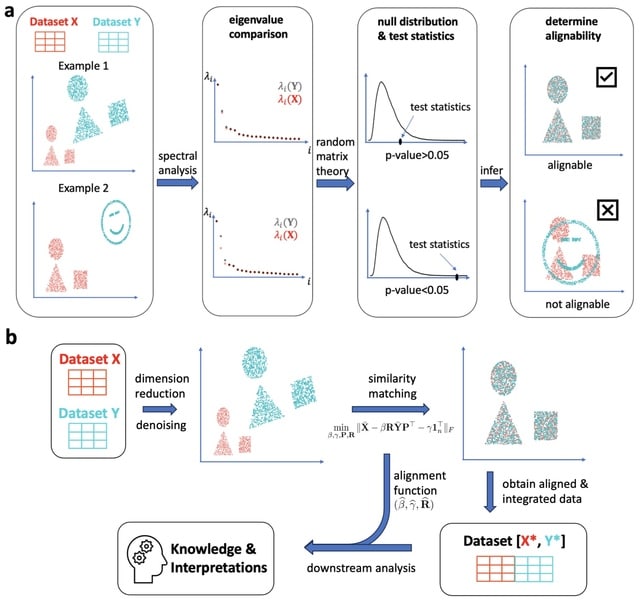
□ SMAI: Is your data alignable? Principled and interpretable alignability testing and integration of single-cell data
>> https://www.biorxiv.org/content/10.1101/2023.08.03.551836v1
SMAI (a spectral manifold alignment and inference) provides a statistical test to robustly determine the alignability between datasets to avoid misleading inference, and is justified by high-dimensional statistical theory. SMAI obtains a symmetric invertible alignment function.
SMAI-align incorporates a high-dimensional shuffled Procrustes analysis, which iteratively searches for the sample correspondence and the best similarity transformation that minimizes the discrepancy between the intrinsic low-dimensional signal structures.
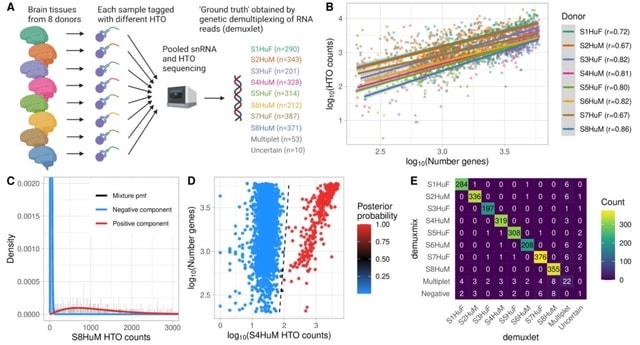
□ demuxmix: Demultiplexing oligonucleotide-barcoded single-cell RNA sequencing data with regression mixture models
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad481/7234612
demuxmix’s probabilistic classification framework provides error probabilities for droplet assignments that can be used to discard uncertain droplets and inform about the quality of the HTO data and the success of the demultiplexing process.
demuxmix utilizes the positive association between detected genes in the RNA library and HTO counts to explain parts of the variance in the HTO data resulting in improved droplet assignments.
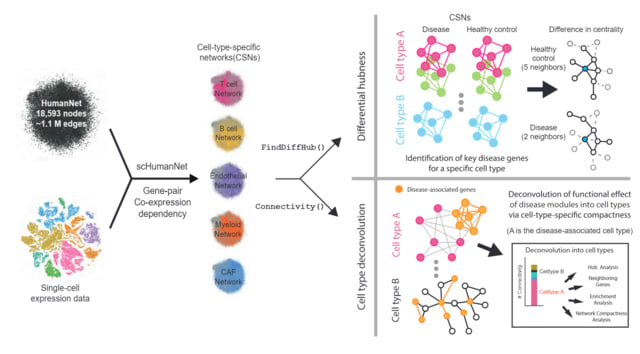
□ scHumanNet: Construction and analysis of cell-type-specific functional gene network, with SCINET and HumanNetv3
>> https://github.com/netbiolab/scHumanNet
scHumanNet enables cell-type specific networks with scRNA-seq data. The SCINET framework takes a single cell gene expression profile and the “reference interactome” HumanNet v3, to construct a list of cell-type specific network.
With the modified version of SCINET source code and the detailed tutorial described below, researchers could take any single-cell RNA sequencing (scRNA-seq) data of any biological context (e.g., disease) and construct their own cell-type specific network for downstream analysis.
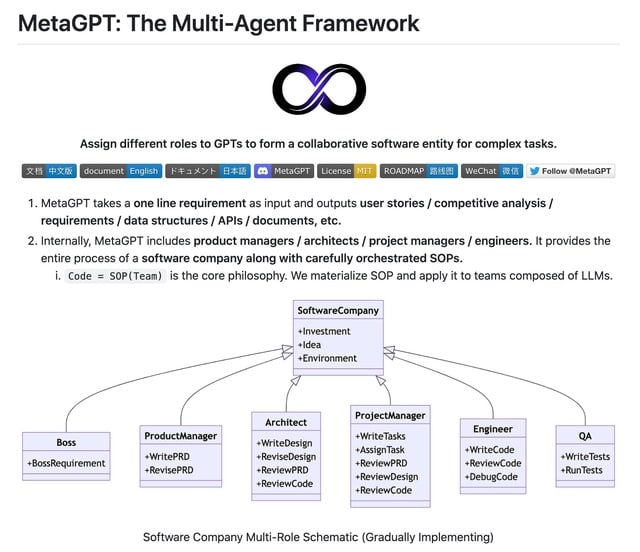
□ Lior RT
>> https://twitter.com/alphasignalai/status/1687878483899207680?s=61&t=YtYFeKCMJNEmL5uKc0oPFg
Impressive. MetaGPT is about to reach 10,000 stars on Github.
It's a Multi-Agent Framework that can behave as an engineer, product manager, architect, project managers.
With a single line of text it can output the entire process of a software company along with carefully orchestrated SOPs:
▸ Data structures
▸ APIs
▸ Documents
▸ User stories
▸ Competitive analysis
▸ Requirements
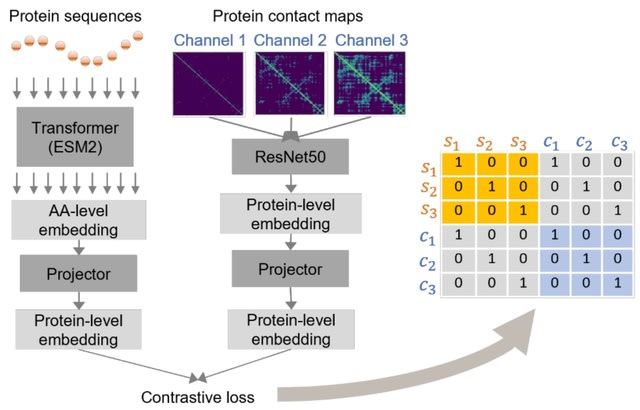
□ S-PLM: Structure-aware Protein Language Model via Contrastive Learning between Sequence and Structure
>> https://www.biorxiv.org/content/10.1101/2023.08.06.552203v1
S-PLM, a 3D structure-aware protein language model developed through multi-view contrastive learning. Unlike the joint-embedding-based methods that rely on both protein structure and sequence for inference, S-PLM encodes the sequence and 3D structure of proteins individually.
S-PLM sequence encoder was fine-tuned based on the pre-trained ESM2 model. S-PLM demonstrates the ability to align sequence and structure embeddings of the same protein effectively while keeping other embeddings from other proteins further apart.

□ Few-shot biomedical named entity recognition via knowledge-guided instance generation and prompt contrastive learning
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btad496/7238215
A knowledge-guided instance generation for few-shot BioNER, which generates diverse and novel entities based on similar semantic relations of neighbor nodes.
And by introducing question prompts, we natively formulate BioNER as a QA task, and propose prompt contrastive learning to improve the robustness of the model by measuring the mutual information between query and entity.

□ The Helix Nebula