□ BioByGANS: biomedical named entity recognition by fusing contextual and syntactic features through graph attention network in node classification framework
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-05051-9
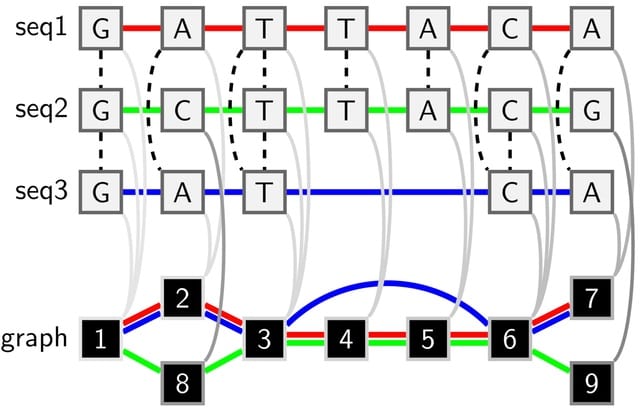
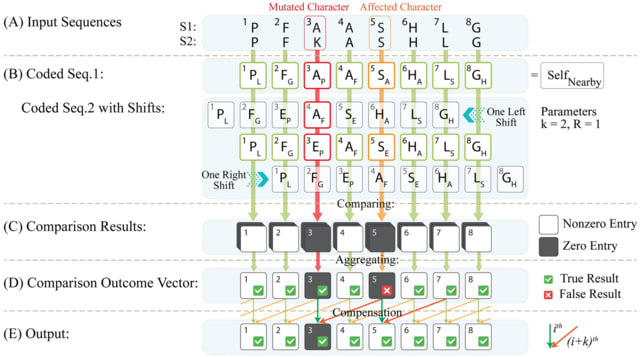
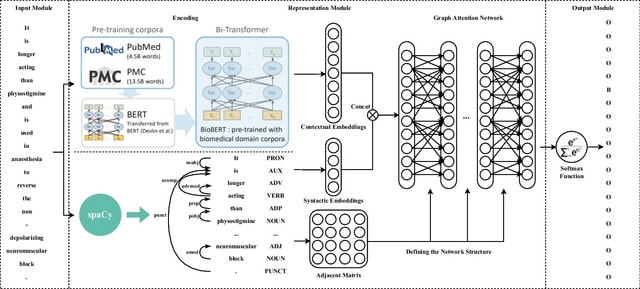
BioByGANS (BioBERT/SpaCy-Graph Attention Network-Softmax) models the dependencies / topology of a sentence and formulate the BioNER task. This formulation can introduce topological features of language and no longer be only concerned about the distance b/n words in the sequence.
First, BioByGANS uses periods to segment sentences and spaces and symbols to segment words. Second, contextual features are encoded by BioBERT, and syntactic features such as part of speeches, dependencies and topology are preprocessed by SpaCy respectively.
A graph attention network is then used to generate a fusing representation considering both the contextual features and syntactic features. Last, a softmax function is used to calculate the probabilities.
□ CARNAGE: Investigating graph neural network for RNA structural embedding
>> https://www.biorxiv.org/content/10.1101/2022.12.02.515916v1
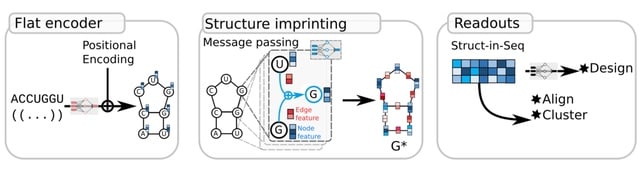
CARNAGE (Clustering/Alignment of RNA with Graph-network Em- bedding), which leverages a graph neural network encoder to imprint structural information into a sequence-like embedding; therefore, downstream sequence analyses now account implicitly for structural constraints.
CARNAGE creates a graphG = (V,E,U), where nodes V are unit-vectors encoding the nucleotide identity. For each node/nucleotide, two rounds of message passing network aggregate information. All the node vectors are concatenated to form the Si-seq.
□ bmVAE: a variational autoencoder method for clustering single-cell mutation data
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac790/6881080
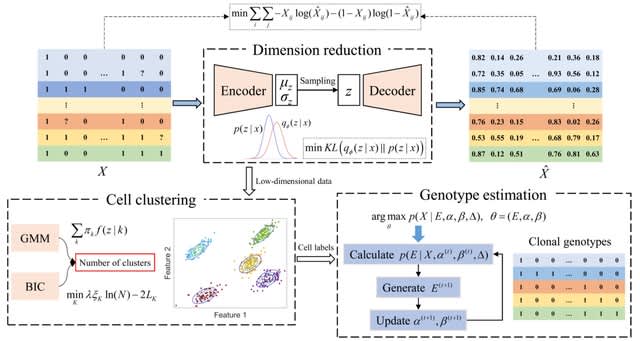
bmVAE infers the low-dimensional representation of each cell by minimizing the Kullback-Leibler divergence loss and reconstruction loss (measured using cross-entropy). bmVAE takes single-cell binary mutation data as inputs, and outputs inferred cell subpopulations as well as their genotypes.
bmVAE employs a VAE model to learn latent representation of each cell in a low-dimensional space, then uses a Gaussian mixture model (GMM) to find clusters of cells, finally uses a Gibbs sampling based approach to estimate genotypes of each subpopulation in the latent space.
□ rcCAE: a convolutional autoencoder based method for detecting tumor clones and copy number alterations from single-cell DNA sequencing data
>> https://www.biorxiv.org/content/10.1101/2022.12.04.519013v1
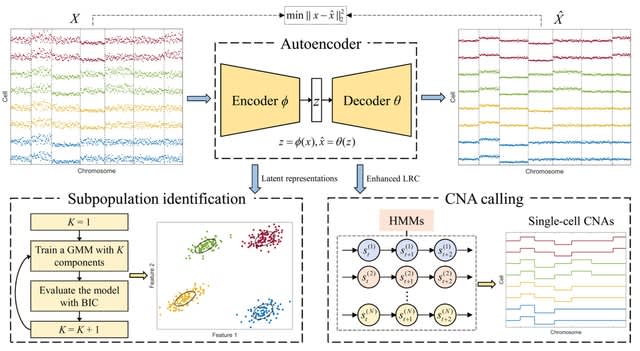
rcCAE uses a convolutional encoder network to project thelog2 transformed read counts (LRC) into a low-dimensional latent space where the cells are clustered into distinct subpopulations through a Gaussian mixture model.
rcCAE leverages a convolutional decoder network to recover the read counts from learned latent representations. rcCAE employs a novel hidden Markov model to jointly segment the genome and infer absolute copy number for each segment.
rcCAE directly deciphers ITH from original read counts, which avoids potential error propagation from copy number analysis to ITH inference. After the algorithm converges, the copy number of each bin is deduced from the state that has the maximum posterior probability.
□ gtexture: Haralick texture analysis for graphs and its application to biological networks
>> https://www.biorxiv.org/content/10.1101/2022.11.21.517417v1
The method for calculating GLCM-equivalents and Haralick texture features and apply it to several network types. They developed the translation of co-occurrence matrix analysis to generic networks for the first time.
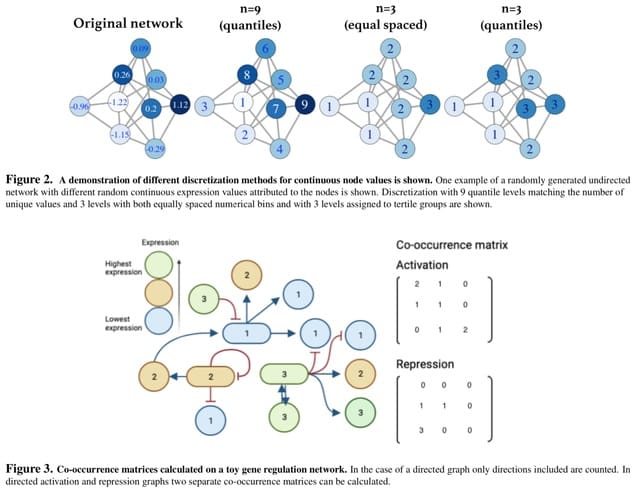
The number of distinct node weights is w, the dimension of the co-occurrence matrix, C, is w × w. Co-occurrence matrices summarize a network when the number of distinct node weights is less than the number of nodes.
gtexture reduces the number of unique node weights, incl. node weight binning options for continuous node weights. Continuous data can be transformed via several discretisation methods.
The Haralick features calculated on different landscapes and networks of the same size but with different topologies vary. Although highly specific methods designed for detecting landscape ruggedness exist, this discretization and co-occurrence matrix method is more generalizable.
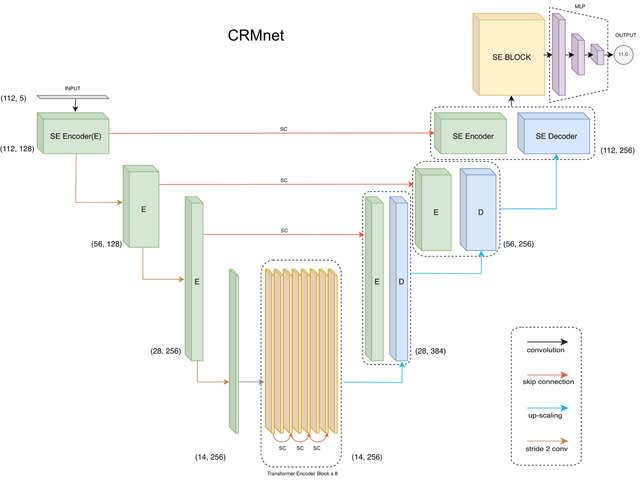
□ CRMnet: a deep learning model for predicting gene expression from large regulatory sequence datasets
>> https://www.biorxiv.org/content/10.1101/2022.12.02.518786v1
CRMnet, a Transformer encoded U-Net from the image semantic segmentation task and applied it to genomic sequences as a feature extractor. CRMnet utilizes transformer encoders, which leverage self-attention mechanisms to extract additional useful information from genomic sequences.
CRMnet consists of Squeeze and Excitation (SE) Encoder Blocks, Transformer Encoder Blocks, SE Decoder Blocks, SE Block and Multi-Layer Perceptron (MLP). CRMnet has an initial encoding stage that extracts feature maps at progressively lower dimensions.
A decoder stage that upscales these feature maps back to the original sequence dimension, whilst concatenating with the higher resolution feature maps of the encoder at each level to retain prior information despite the sparse upscaling.
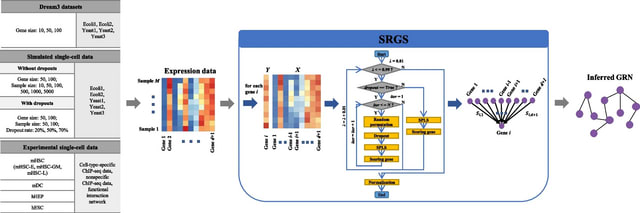
□ SRGS: sparse partial least squares-based recursive gene selection for gene regulatory network inference
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-09020-7
SRGS, SPLS (sparse partial least squares)-based recursive gene selection, to infer GRNs from bulk or single-cell expression data. SRGS recursively selects and scores the genes which may have regulations on the considered target gene based on SPLS.
SRGS recursively selects and scores the genes which may have regulations on the considered target gene. They randomly scramble samples, set some values in the expression matrix to zeroes, and generate multiple copies of data through multiple iterations.
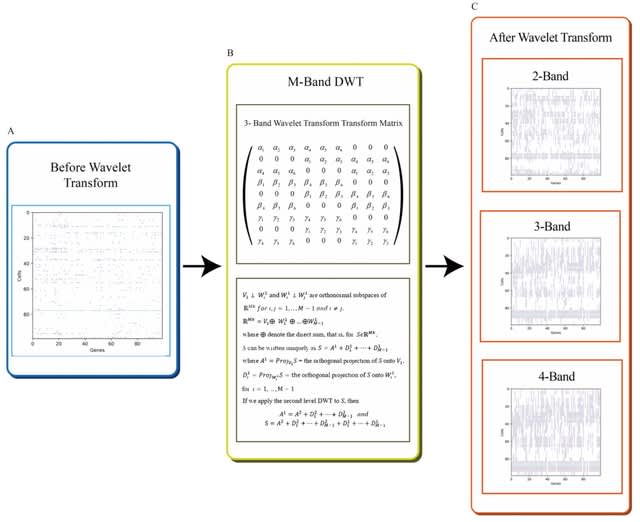
□ WINC: M-Band Wavelet-Based Imputation of scRNA-seq Matrix and Multi-view Clustering of Cell
>> https://www.biorxiv.org/content/10.1101/2022.12.05.519090v1
WINC integrates M-band wavelet analysis and UMAP to a panel of single cell sequencing datasets via breaking up the data matrix into a trend (low frequency or low resolution) component and (M − 1) fluctuation (high frequency or high resolution) components.
This strategy resolves the notorious chaotic sparsity of droplet RNA-Seq matrix and uncovers missed / rare cell types, identities, states. A non-parametric wavelet-based imputation algorithm of sparse data that integrates M-band orthogonal wavelet for recovering dropout events.

□ DeepPHiC: Predicting promoter-centered chromatin interactions using a novel deep learning approach
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac801/6887158
DeepPHiC adopts a “shared knowledge transfer” strategy for training the multi-task learning model. When tissue A/B is of interest, and aggregates all chromatin interactions from other tissues except tissue A/B to pretrain the shared feature extractor.
DeepPHiC consists of three types of input features, which include genomic sequence and epigenetic signal in the anchors as well as anchor distance. DeepPHiC uses one-hot encoding for the genomic sequence. As a result, the genomic sequence is converted into a 2000 × 4 matrix.
The network architecture of DeepPHiC is developed based on the DenseNet. DeepPHiC uses a ResNet-style structure with skip connections. During back propagation, each layer has a direct access to the output gradients, resulting in faster network convergence.
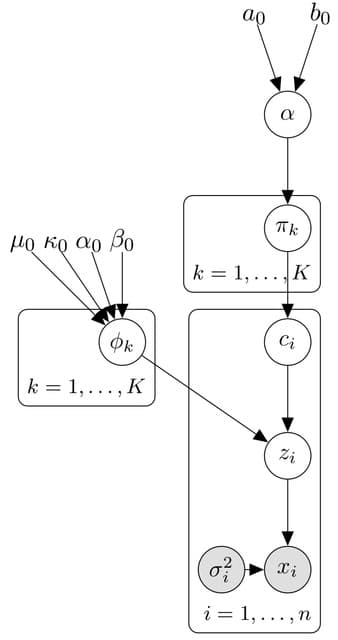
□ DPMUnc: Bayesian clustering with uncertain data
>> https://www.biorxiv.org/content/10.1101/2022.12.07.519476v1
Dirichlet Process Mixtures with Uncertainty (DPMUnc), an extension of a Bayesian nonparametric clustering algorithm which makes use of the uncertainty associated with data points.
DPMUnc outperformed its comparators kmeans and mclust by a small margin when observation noise and cluster variance were small, which increased with increasing cluster variance or observation noise.
DPMZeroUnc is the adjusted version of the datasets where the uncertainty estimates were shrunk to 0. The latent variables are essentially fixed to be equal to the observed data points throughout.
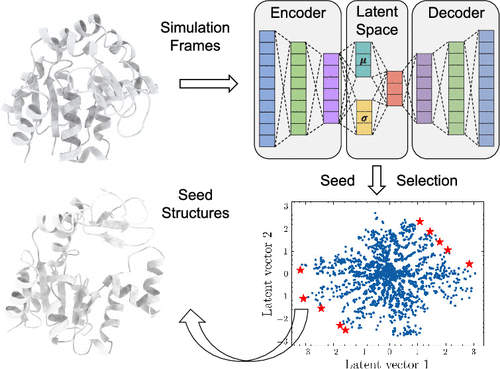
□ LAST: Latent Space-Assisted Adaptive Sampling for Protein Trajectories
>> https://pubs.acs.org/doi/10.1021/acs.jcim.2c01213
LAST accelerates the exploration of protein conformational space. This method comprises cycles of (i) variational autoencoder training, (ii) seed structure selection on the latent space, and (iii) conformational sampling through additional Molecular dynamics simulations.
In metastable ADK simulations, LAST explored two transition paths toward two stable states, while SDS explored only one and cMD neither. In VVD light state simulations, LAST was three times faster than cMD simulation with a similar conformational space.
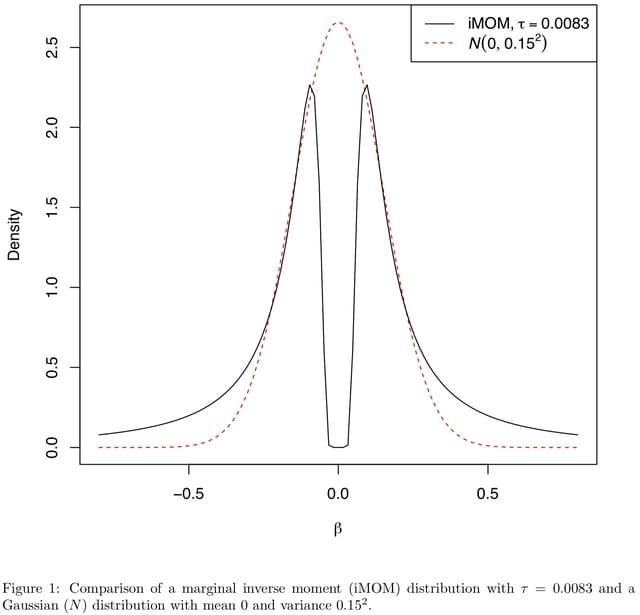
□ FiniMOM: Genetic fine-mapping from summary data using a non-local prior improves detection of multiple causal variants
>> https://www.biorxiv.org/content/10.1101/2022.12.02.518898v1
FiniMOM (fine-mapping using a product inverse-moment priors), a novel Bayesian fine-mapping method for summarized genetic associations. The method uses a non-local inverse-moment prior, which is a natural prior distribution to model non-null effects in finite samples.
FiniMOM allows a non-zero probability for all variables, instead of considering only the variables that correlate highly with the residuals of the current model.
FiniMOM’s sampling scheme is related to reversible jump MCMC algorithm, however this formulation and use of Laplace’s method avoids complicated sampling from varying-dimensional model space.
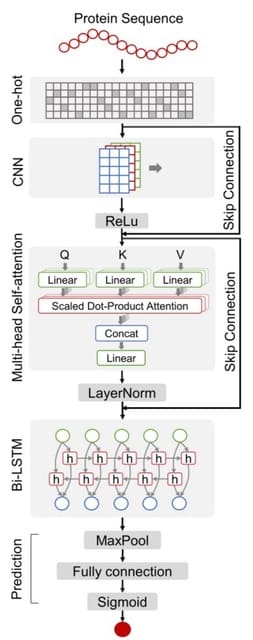
□ DeepCellEss: Cell line-specific essential protein prediction with attention-based interpretable deep learning
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac779/6865030
DeepCellEss utilizes convolutional neural network and bidirectional long short-term memory to learn short- and long-range latent information from protein sequences. Further, a multi-head self-attention mechanism is used to provide residue-level model interpretability.
DeepCellEss converts a protein sequence into a numerical matrix using one-hot encoding. The multi-head self-attention is used to produce residue-level attention scores. After this, a bi-LSTM module is applied to model sequential data by learning long-range dependencies.
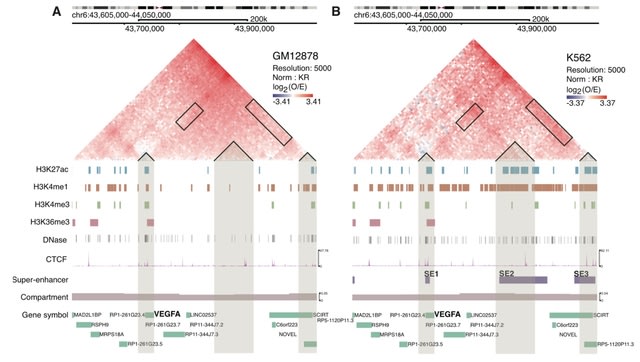
□ DiffDomain enables identification of structurally reorganized topologically associating domains
>> https://www.biorxiv.org/content/10.1101/2022.12.05.519135v1
DiffDomain, an algorithm leveraging high-dimensional random matrix theory to identify structurally reorganized TADs using chromatin contact maps. DiffDomain outperforms alternative methods for FPRs, TPRs, and identifying a new subtype of reorganized TADs.
DiffDomain directly computes a difference matrix then normalize it properly, skipping the challenging normalization steps for individual Hi-C contact matrices. DiffDomain then borrows well-established theorectical results in ramdom matrix theory to compute a theorectical P value.
DiffDomain identifies reorganized TADs b/n cell types w/ reasonable reproducibility using pseudo-bulk Hi-C data from as few as 100 cells per condition. DiffDomain reveals that TADs have clear differential cell-to-population variability and heterogeneous cell-to-cell variability.

□ Efficient inference and identifiability analysis for differential equation models with random parameters
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010734
A new likelihood-based framework, based on moment matching, for inference and identifiability analysis of differential equation models that capture biological heterogeneity through parameters that vary according to probability distributions.
The availability of a surrogate likelihood allows us to perform inference and identifiability analysis of random parameter models using the standard suite of tools, including profile likelihood, Fisher information, and Markov-chain Monte-Carlo.

□ EDIR: Exome Database of Interspersed Repeats
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac771/6858440
The Exome Database of Interspersed Repeats (EDIR) was developed to provide an overview of the positions of repetitive structures within the human genome composed of interspersed repeats encompassing a coding sequence.
EDIR can be queried for interspersed repeat sequence IRS in a gene of interest. Additional parameters which can be entered are the length of the repeat (7-20 bp), the minimum (0 bp) and maximum distance (1000 bp) of the spacer sequence, and whether to allow a 1-bp mismatch.
As output, a table is given where for each repeat length, the number of interspersed repeat structures, together with the average distance separating two repeats, as well as the number of interspersed repeat structures per megabase and whether a 1 bp mismatch has occurred.
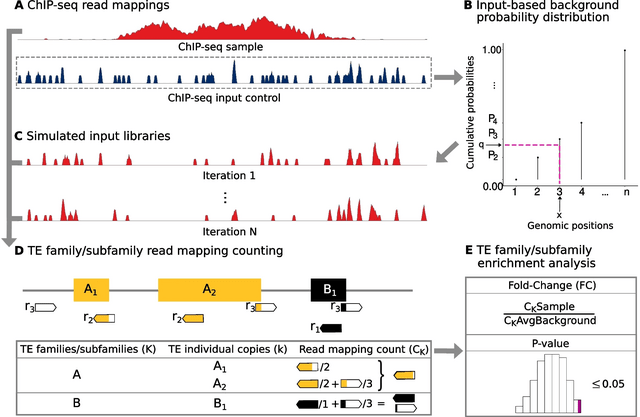
□ T3E: a tool for characterising the epigenetic profile of transposable elements using ChIP-seq data
>> https://mobilednajournal.biomedcentral.com/articles/10.1186/s13100-022-00285-z
The Transposable Element Enrichment Estimator (T3E) weights the number of read mappings assigned to the individual TE copies of a family/subfamily by the overall number of genomic loci to which the corresponding reads map, and this is done at the single nucleotide level.
T3E maps ChIP-seq reads to the entire genome of interest w/o subsequently remapping the reads to particular consensus or pseudogenome sequences. In its calculations T3E considers the number of both repetitive / non-repetitive genomic loci to which each multimapper mapped.
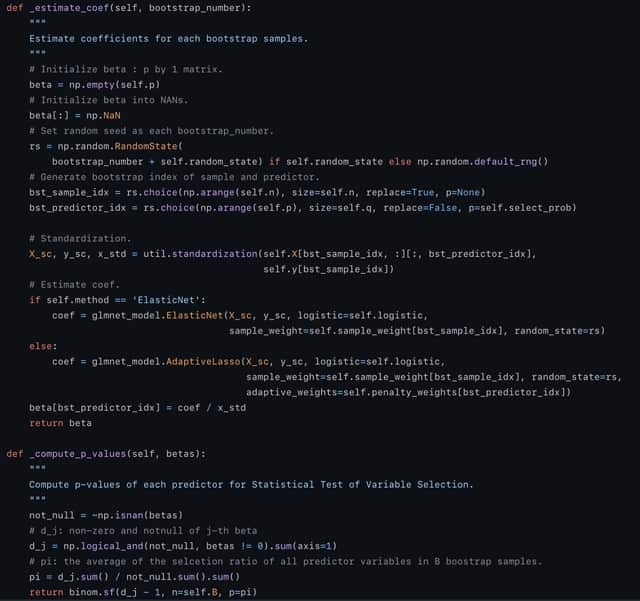
□ Hi-LASSO: High-performance python and apache spark packages for feature selection with high-dimensional data
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0278570
Random LASSO does not take advantage of global oracle property. Although Random LASSO uses bootstrapping with weights being proportional to importance scores of predictors in the second procedure, the final coefficients are estimated without the weights.
Hi-LASSO computes importance scores of variables by averaging absolute coefficients. Hi-LASSO alleviates bias from bootstrapping, improves the performance taking advantage of global oracle property, provides a statistical strategy to determine the number of bootstrapping.
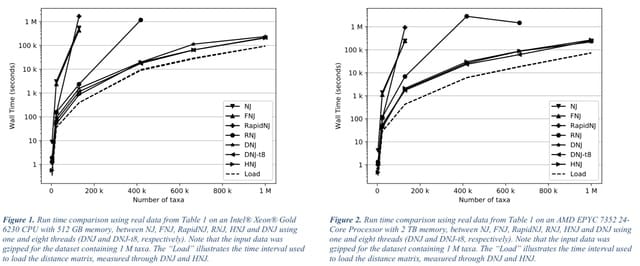
□ Scaling Neighbor-Joining to One Million Taxa with Dynamic and Heuristic Neighbor-Joining
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac774/6858462
Dynamic and Heuristic Neighbor-Joining, are presented, which optimize the canonical Neighbor-Joining method to scale to millions of taxa without increasing the memory requirements.
Both Dynamic and Heuristic Neighbor-Joining outperform the current gold standard methods to construct Neighbor-Joining trees, while Dynamic Neighbor-Joining is guaranteed to produce exact Neighbor-Joining trees.
Asymptotically, DNJ reaches a runtime of O(n3) when updates to D causes frequent updates. This worst-case time complexity can be reduced to O(n2) with an approximating search heuristic. The time complexity of HNJ to O(n2), while the space complexity remains at O(n2) as for DNJ.
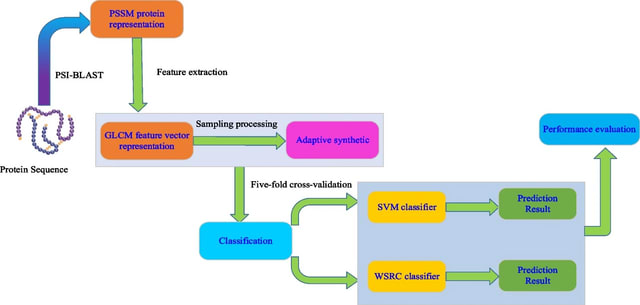
□ GLCM-WSRC: Robust and accurate prediction of self-interacting proteins from protein sequence information by exploiting weighted sparse representation based classifier
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04880-y
GLCM-WSRC (gray level co-occurrence matrix-weighted sparse representation based classification), for predicting SIPs automatically based on protein evolutionary information from protein primary sequences.
The GLCM algorithm is employed to capture the valuable information from the PSSMs and form feature vectors, after which the ADASYN is applied to balance the training data set to form new feature vectors used as the input of classifier from the GLCM feature vectors.

□ Treenome Browser: co-visualization of enormous phylogenies and millions of genomes
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac772/6858450
Treenome Browser displays mutations as vertical lines spanning the mutation’s presence among samples in the phylogeny, drawn at their horizontal position in an associated reference genome.
The core algorithm used by Treenome Browser decodes a mutation-annotated tree to compute the on-screen position of each mutation in the tree. To compute vertical positions, the vertical span of each subclade of the tree is first stored using dynamic programming.
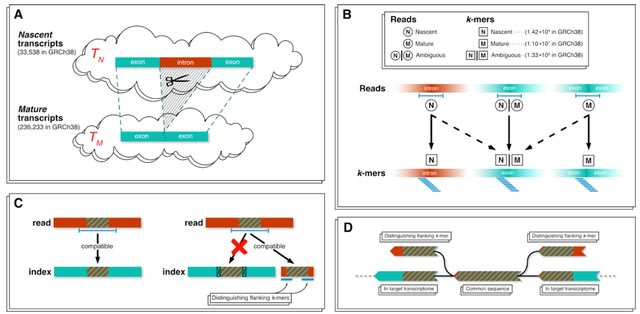
□ Accurate quantification of single-nucleus and single-cell RNA-seq transcripts
>> https://www.biorxiv.org/content/10.1101/2022.12.02.518832v1
The presence of both nascent and mature mRNA molecules in single-cell RNA-seq data leads to ambiguity in the notion of a “count matrix”. Underlying this ambiguity, is the challenging problem of separately quantifying nascent and mature mRNAs.
By utilizing k-mers, this approach has the benefit of being efficient as it is compatible with pseudoalignment. An approach to quantification of single-nucleus RNA-seq that focuses on the nascent transcripts, thereby mirroring the approach that focuses on mature transcripts.
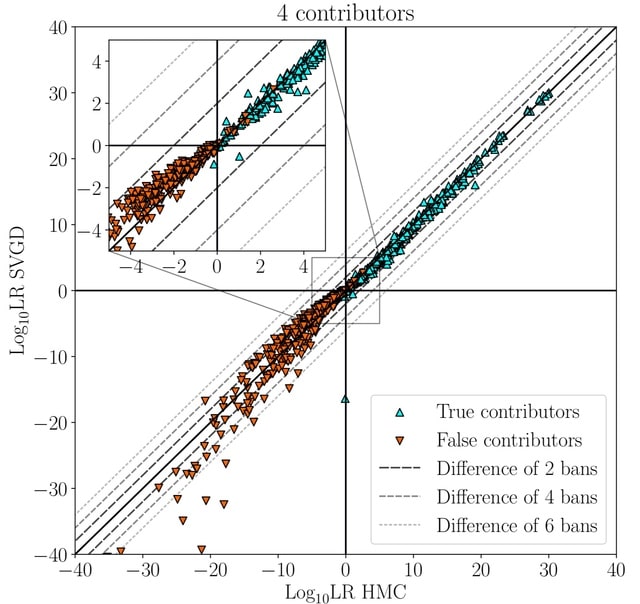
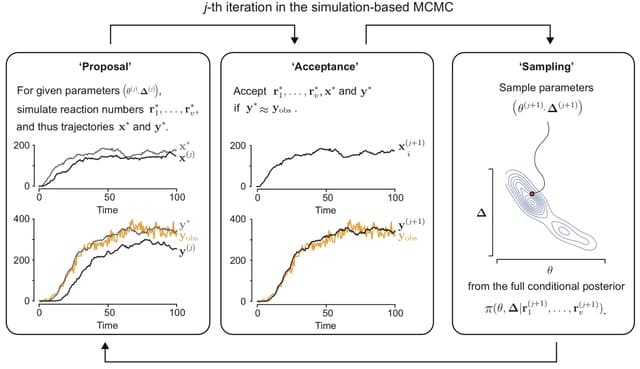
□ Variational inference accelerates accurate DNA mixture deconvolution
>> https://www.biorxiv.org/content/10.1101/2022.12.01.518640v1
Considering Stein Variational Gradient Descent (SVGD) and Variational Inference (VI) with an evidence lower-bound objective. Both provide alternatives to the commonly used Markov-Chain Monte-Carlo methods for estimating the model posterior in Bayesian probabilistic genotyping.
The model defines the unnormalised posterior, and the estimator defines the way how an approximation of this distribution is obtained. These two parts are largely independent of each other, meaning that, for example, an estimator can be replaced with another one.
The singularities are not a problem for HMC estimators, who will avoid them
because of the high curvature of the posterior in the vicinity of the singularities. The trajectory of the simulated Hamiltonian differs too much from the expected Hamiltonian.

□ HTRX: an R package for learning non-contiguous haplotypes associated with a phenotype
>> https://www.biorxiv.org/content/10.1101/2022.11.29.518395v1
HTRX defines a template for each haplotype using the combination of ‘0’, ‘1’ and ‘X’ which represent the reference allele, alternative allele and either of the alleles, at each SNP. A four-SNP haplotype ‘1XX0’ only refers to the interaction between the first and the fourth SNP.
HTRX considers lasso penalisation. AIC and BIC penalise the number of features through forward regression, and the features whose parameters do not shrink to 0 are retained. The objective function of HTRX is the out-of-sample variance explained by haplotypes within a region.
□ GSSNNG: Gene Set Scoring on the Nearest Neighbor Graph (gssnng) for Single Cell RNA-seq (scRNA-seq)
>> https://www.biorxiv.org/content/10.1101/2022.11.29.518384v1
GSSNNG produces a gene set score for each individual cell, addressing problems of low read counts and the many zeros and retains gradations that remain visible in UMAP plots.
The method works by using a nearest neighbor graph in gene expression space to smooth the count matrix. The smoothed expression profiles are then used in single sample gene set scoring calculations.
Using gssnng, large collections of cells can be scored quickly even on a modest desktop. The method uses the nearest neighbor graph (kNN) of cells to smooth the gene expression count matrix which decreases sparsity and improves geneset scoring.
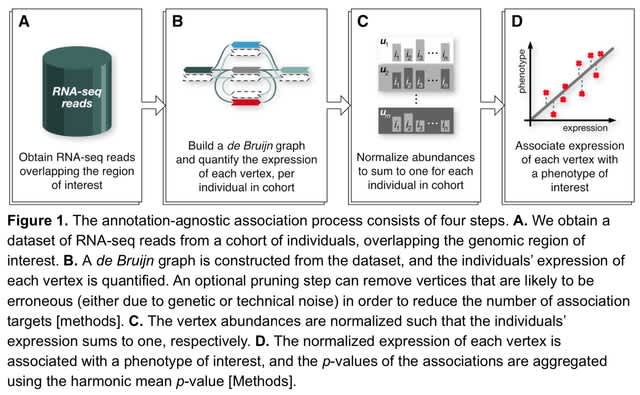
□ Annotation-agnostic discovery of associations between novel gene isoforms and phenotypes
>> https://www.biorxiv.org/content/10.1101/2022.12.02.518787v1
A bi-directed de Bruijn Graph (dBG) is constructed, using Bifrost, from these reads with k-mer size 𝑘 = 31 and then compacted such that consecutive k-mers with out-degree 1 and in-degree 1 respectively are folded into a single, maximal unitig, which is a high-confidence contig.
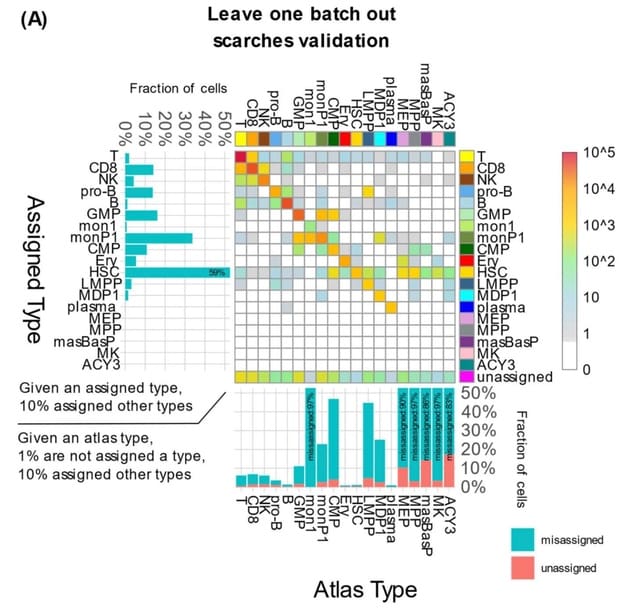
□ MCProj: Metacell projection for interpretable and quantitative use of transcriptional atlases
>> https://www.biorxiv.org/content/10.1101/2022.12.01.518678v1
MCProj, an algorithm for quantitative analysis of query scRNA-seq given a reference atlas. The algorithm is transforming single cells to quantitative states using a metacell representation of the atlas and the query.
MCProj infers each query state as a mixture of atlas states, and tags cases in which such inference is imprecise, suggestive of novel or noisy states in the query. MCProj tags novel query states and compares them to atlas states.
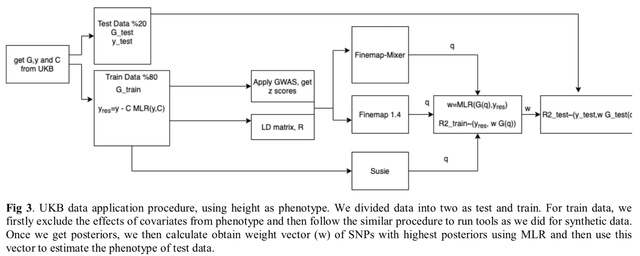
□ Finemap-MiXeR: A variational Bayesian approach for genetic finemapping
>> https://www.biorxiv.org/content/10.1101/2022.11.30.518509v1
The Finemap-MiXeR is based on a variational Bayesian approach for finemapping genomic data, i.e., determining the causal SNPs associated with a trait at a given locus after controlling for correlation among genetic variants due to linkage disequilibrium.
Finemap-MiXeR on the optimization of Evidence Lower Bound of the likelihood function obtained from the MiXeR model. The optimization is done using Adaptive Moment Estimation Algorithm, allowing to obtain posterior probability of each SNP to be a causal variant.
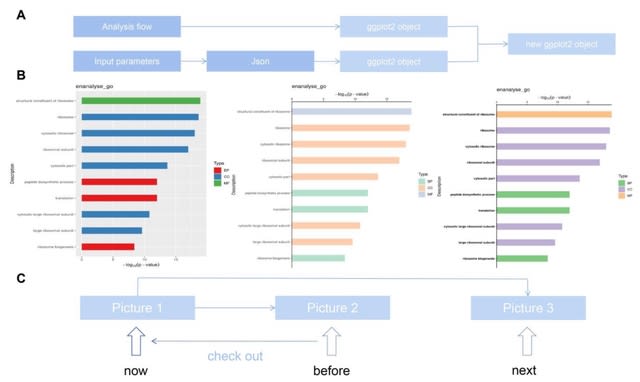
□ Visual Omics: A web-based platform for omics data analysis and visualization with rich graph-tuning capabilities
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac777/6865031
Visual Omics integrates multiple omics analyses which include differential expression analysis, enrichment analysis, protein domain prediction and protein-protein interaction analysis with extensive graph presentations.
The extensive use of the powerful downstream ggplot2 and its family packages enables almost all analysis results to be visualized by Visual Omics and can be adapted to the online tuning system almost without modification.
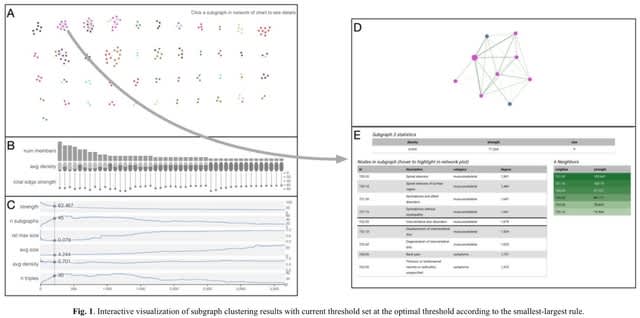
□ associationSubgraphs: Interactive network-based clustering and investigation of multimorbidity association matrices
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac780/6874541
associationSubgraphs, a new interactive visualization method to quickly and intuitively explore high-dimensional association datasets using network percolation and clustering.
The algorithm for computing associationSubgraphs at all given cutoffs is closely related to single-linkage clustering but differs philosophically by viewing nodes that are yet to be merged with other nodes as unclustered rather than residing within their own cluster of size one.
It investigates association subgraphs efficiently, each containing a subset of variables with more frequent associations than the remaining variables outside the subset, by showing the entire clustering dynamics and provide subgraphs under all possible cutoff values at once.