









遺伝子分野に限らず、技術工学の取り得る選択肢で『倫理的に問題』であるとはどういうことか。個々の行動規範が、議論やコンセンサスを経た結果であるかどうかは、単純に予測と結果の仮説の上にのみ成り立つ。この問題を制御する上で本当に必要なのは、技術と力学的関係にある社会規範の検証である。可能なことを実証し続けなければ、我々に待っているのは死滅の運命だ。遺伝子工学がカタストロフィに向かうかどうかは、個々が然るべきトランザクションを経たとしても予測不可能である。
過ってはいけないのが、生命としての動機だ。感情は思考を定め、思考は行動を決定する。そこにはあるのは、自己と非自己の振る舞いの差異だけである。生命種は、相互作用する振動子の共振のレベルと摂動関係に置き換えられる。振る舞いに干渉するのであれば、別の記号でも知性でも人工知能でも問題ではない。このアルゴリズムの瑕疵はシステムの膜の外側に向かって、予め決定論的に発現する。我々は過ちを侵すが、侵さなければ エラーを修正できない。
□ PEAS: A neural network based model effectively predicts enhancers from clinical ATAC-seq samples:
>> https://www.nature.com/articles/s41598-018-34420-9
Among the tools developed by the ENCODE consortium, the Hidden Markov Model (HMM)–based ChromHMM algorithm has become an important tool to assess the global epigenomic landscape in human cells by segmenting genome-wide chromatin into a finite number of chromatin states. Although ChromHMM has been very powerful in finding regulatory elements in diverse human cell types, ChromHMM cannot be applied on clinical samples since the datasets that it stem from (i.e., multiple ChIP-seq profiles) cannot be easily generated in these samples.
□ Clustering-based optimization method of reference set selection for improved CNV callers performance:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/25/478313.1.full.pdf
CODEX algorithm is based on a multi-sample normalization model, which is fitted to remove various biases including noise introduced by different GC content in the analyzed targets, and CNVs are called by the Poisson likelihood-based segmentation algorithm. ExomeCopy implements a hidden Markov model which uses positional covariates, including background read depth and GC content, to simultaneously normalize and segment the samples into the regions of constant copy count.
□ "on the definition of sequence identity":
>> http://lh3.github.io/2018/11/25/on-the-definition-of-sequence-identity
estimate error rate or identity:
minimap2 -c ref.fa query.fa \
| perl -ane 'if(/tp:A:P/&&/NM:i:(\d+)/){$n+=$1;$m+=$1 while/(\d+)M/g;$g+=$1,++$o while/(\d+)[ID]/g}END{print(($n-$g+$o)/($m+$o),"\n")}'
"The estimate of sequence identity varies with definitions and alignment scoring. When you see someone talking about “sequencing error rate” next time, ask about the definition and scoring in use to make sure that is the error rate you intend to compare."
□ StructLMM: A linear mixed-model approach to study multivariate gene–environment interactions:
>> https://www.nature.com/articles/s41588-018-0271-0
Although high-dimensional environmental data are increasingly available and multiple exposures have been implicated with G×E at the same loci, multi-environment tests for G×E are not established. while StructLMM can in principle be used in conjunction with any environmental covariance, they have here limited the application to linear covariances. The model could be extended to account for non-linear interactions, for example using polynomial covariance functions.
□ Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions:
>> https://www.nature.com/articles/s41588-018-0268-8
This flexible approach increases power, improves effect estimates and allows for more quantitative assessments of effect-size heterogeneity compared to simple shared or condition-specific assessments. although genetic effects on expression are extensively shared among tissues, effect sizes can still vary greatly among tissues. Some shared eQTLs show stronger effects in subsets of biologically related tissues, or in only one tissue (for example, testis).
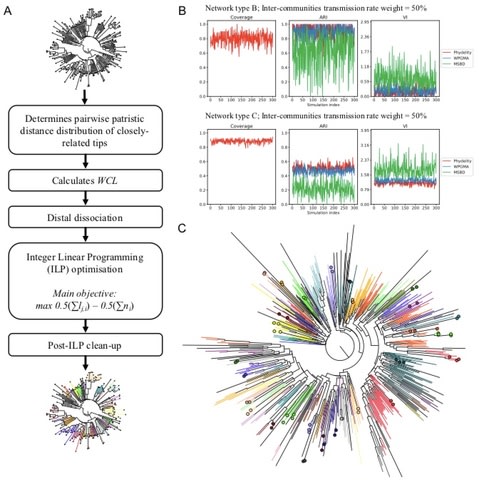
□ Inferring putative transmission clusters with Phydelity:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/26/477653.full.pdf
Phydelity identifies groups of sequences that are more closely-related than the ensemble distribution of the phylogeny under a statistically-principled and phylogeny-informed framework, without the introduction of arbitrary distance thresholds. Phydelity infers the within-cluster divergence of putative transmission clusters by first determining the pairwise patristic distance distribution of closely-related tips. In simulated phylogenies, Phydelity achieves higher rates of correspondence to ground-truth clusters than current model-based methods, and comparable results to parametric methods without the need for parameter calibration.
□ Algorithm identifies multiple gene–environment relationships:
>> https://www.ebi.ac.uk/about/news/press-releases/gene-environment-algorithm
Comprehensive analysis of hundreds of environmental factors could enhance understanding of genotype–phenotype relationships

□ Using classification algorithms, such as support vector machines and neural networks, to automatically find efficient linear and non-linear collective variables for accelerated molecular simulations:
>> https://aip.scitation.org/doi/10.1063/1.5029972
solving the “initial” CV problem using a data-driven approach inspired by the field of supervised machine learning (SML). In particular, they show how the decision functions in SML algorithms can be used as initial CVs (SMLcv) for accelerated sampling. they illustrate how the distance to the support vector machines’ decision hyperplane, the output probability estimates from logistic regression, shallow or deep neural network classifiers, and other classifiers may be used to reversibly sample slow structural transitions.
□ SLEDGE Hammer: Swift Large-scale Examination of Directed Genome Editing:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/27/479261.full.pdf
The robust isolation and detection of multiple alleles of various abundancies in a mosaic genetic background allows phenotype-genotype correlation already in the injected generation, demonstrating the reliability and sensitivity of the filter-in-tips. the SLEDGE Hammer protocol with the adapted filter-in pipet tips, allows to bypass the otherwise tedious and time-consuming genomic purification step that hitherto limited high-throughput genotyping approaches.
□ OPTIMIR: a novel algorithm for integrating available genome- wide genotype data into miRNA sequence alignment analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/27/479097.full.pdf
OPTIMIR, for pOlymorPhisminTegratIon for MIRna data alignment, is based on a scoring strategy that incorporate biological knowledge on miRNA editing to identify the most likely alignment in presence of cross-mapping reads. OPTIMIR integrates genetic information from genotyping arrays or DNA sequencing into the miRSeq data alignment process with the aim of improving the accuracy of polymiRs alignment, while accommodating for isomiRs detection and ambiguously aligned reads.

□ MinION Mk 1c will combine MinION, MinIT for rapid data analysis and a screen, for a one-stop palm sized fully portable sequencing system, fully connected.
Mk1C has mobile/cellular data, NVMe SSD built-in

□ PromethION is now giving us more than a Tb of @nanopore sequencing data per week. This is a ‘world changing phenomenon’#nanoporeconf

□ algorithms for SV detection by while genome alignment: RaGOO beats SALSA #nanoporeconf
□ Hidden Markov Models (HMMs) not only for calling signal, DNA storage can use paired HMM state machines; takes binary data, coverts to ternary data then encodes as DNA. Further extension to this - @nanopore DNA seq can be used for protein alignments #nanoporeconf

□ the cost calculation of RAGE sequencing SCISOR-seq / RAGE-seq
□ initial strategy was a mix of short, long, and linked reads. Encouraging results on PromethION motivated a switch to solely using PromethION. Currently running 6-8 flow cells in parallel twice a week, >1 terabase per week. "This is a world-changing phenomenon" #NanoporeConf

□ The @nanopore PromethION 48fc, with a theoretical maximum throughput of 15Tb per 48 fcell run, or 5769Gb per day, is now on par with the announced but not released @MGI_BGI T7 machine (not available until Q2/Q3 2019)
□ CoCo: RNA-seq Read Assignment Correction for Nested Genes and Multimapped Reads:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/29/477869.full.pdf
a correction of the gene annotation used by read assignment softwares such as featureCounts or HTSeq in order to correct the evaluated read counts for embedded genes such as snoRNA, that overlap features of their host gene's transcripts such as retained introns and exons. The second part of the correction distributes multi mapped reads in relation to the evaluated read counts obtained from single mapped reads. CoCo salvages over 15% of discarded aligned RNA-seq reads and significantly changes the abundance estimates for both coding and non-coding RNA as validated by PCR and bed-graph comparisons.
□ A Bayesian mixture modelling approach for spatial proteomics:
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006516
a Bayesian generative classifier based on Gaussian mixture models to assign proteins probabilistically to sub-cellular niches, proteins have a probability distribution over sub-cellular locations, using the expectation-maximisation algorithm, as well as Markov-chain Monte-Carlo. Outliers are often dispersed and thus this additional component is described by a heavy-tailed distribution: the multivariate Student’s t-distribution, leading us to a T-Augmented Gaussian Mixture model (TAGM).
□ Limits to a classic paradigm: Most transcription factors regulate genes in multiple biological processes:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/28/479857.full.pdf
In this scenario, general regulons show the regulatory potential of TFs, but the specific subset of genes in the regulon that is expressed at a certain time is defined by the combinatory logic of the TFs bound to each gene’s promoter. Dissecting the molecular decision-making processes associated to changes of growth conditions at a genomic level is doable with current technologies.
□ GRAM: A generalized model to predict the molecular effect of a non-coding variant in a cell-type specific manner:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/29/482992.full.pdf
using a LASSO regularized linear model, transcription factor binding most predictive, especially for TFs that are hubs in the regulatory network; in contrast, evolutionary conservation, a popular feature in many other functional-impact predictors, has almost no contribution. Moreover, TF binding inferred from in vitro SELEX is as effective as that from in vivo ChIP-Seq. Second, we implemented GRAM integrating SELEX features and expression profiles. The GRAM model will be a useful tool for elucidating the underlying patterns of variants that modulate expression in a cell-type context. By leveraging the accumulating data generated from multiple cell lines, future studies can be performed in-depth investigation using GRAM.
□ FastqPuri: high-performance preprocessing of RNA-seq data:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/29/480707.full.pdf
FastqPuri provides sequence quality reports on the sample and dataset level with new plots which facilitate decision making for subsequent quality filtering. Using the BLOOM method to filter out potential contaminations using larger-sized files (e.g. genomes), FastqPuri was faster than BioBloom tools in generating the bloom filter but slightly slower in classifying sequences.
□ scGen: Generative modeling and latent space arithmetics predict single-cell perturbation response across cell types, studies and species:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/29/478503.full.pdf
scGen, a model combining variational autoencoders and latent space vector arithmetics for high-dimensional single-cell GE data. scGen learns cell type and species specific response implying that it captures features that distinguish responding from non-responding genes and cells. By adequately encoding the original expression space in a latent space, achieve simple, near-to-linear mappings for highly non-linear sources of variation in the original data, which explain a large portion of the variability in the data.
□ CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1590-2
The new database contains 42,611 genes, of which 20,352 are potentially protein-coding and 22,259 are noncoding, and a total of 323,258 transcripts. These include 224 novel protein-coding genes and 116,156 novel transcripts. detected over 30 million additional transcripts at more than 650,000 genomic loci, nearly all of which are likely nonfunctional, revealing a heretofore unappreciated amount of transcriptional noise in human cells.
the novel genes and transcripts in CHESS using a genome-guided assembly pipeline including HISAT2 and StringTie. All of these samples were subjected to deep RNA-sequencing, with tens of millions of sequences (“reads”) captured from each sample.
□ NucBreak: Location of structural errors in a genome assembly by using paired-end Illumina reads:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/29/393488.full.pdf
NucBreak aimed at detecting structural errors in assemblies, including insertions, deletions, duplications, inversions, and different inter- and intra-chromosomal rearrangements. NucBreak analyses the alignments of reads properly mapped to an assembly and exploits information about the alternative read alignments.
compared NucBreak with other existing assembly accuracy assessment tools, namely Pilon, REAPR, and FRCbam as well as with several structural variant detection tools, including BreakDancer, Lumpy, and Wham, by using both simulated and real datasets. The benchmarking results have shown that NucBreak in general predicts assembly errors of different types and sizes with relatively high sensitivity and with higher precision than the other tools.

□ NucMerge: Genome assembly quality improvement assisted by alternative assemblies and paired-end Illumina reads:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/30/483701.full.pdf
The tool corrects insertion, deletion, substitution, and inversion errors and locates different inter- and intra-chromosomal rearrangement errors. NucMerge was compared to two existing alternatives, namely Metassembler and GAM-NGS. The results have shown that the error detection approach used in NucMerge is more effective than the CE-statistics and depth-of-coverage analysis.
□ Random Tanglegram Partitions (Random TaPas): An Alexandrian Approach to the Cophylogenetic Gordian Knot:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/29/481846.full.pdf
Random Tanglegram Partitions (Random TaPas) that applies a given global-fit method to random partial tanglegrams of a fixed size to identify the associations, terminals and nodes that maximize phylogenetic congruence. In addition, with time-calibrated trees, Random TaPas is also efficient at distinguishing cospeciation from pseudocospeciation. Random TaPas can handle large tanglegrams in affordable computational time and incorporates phylogenetic uncertainty in the analyses.
□ Linked-read sequencing of gametes allows efficient genome-wide analysis of meiotic recombination:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/30/484022.full.pdf
a highly efficient method for genome-wide identification of COs at kilobase resolution in pooled recombinants. The simplicity of this approach now enables the simultaneous generation and analysis of multiple CO landscapes and thereby allows for efficient comparison of genotypic and environmental effects on recombination, accelerating the pace at which the mechanisms for the regulation of recombination can be elucidated.

□ TorchCraftAI and CherryPi: a machine learning model for high-level strategy selection
>> https://torchcraft.github.io/TorchCraftAI/blog/2018/11/28/build-order-switch-retraining-has-arrived.html
TorchCraftAI: distributed RL environment.
CherryPI: modular StarCraft bot with a hybrid architecture combining rules/search and deep learning.
□ PoreOver: Nanopore basecalling in TensorFlow:
>> https://github.com/jordisr/poreover
PoreOver is a neural network basecaller for the Oxford Nanopore sequencing platform and is under active development. It is intended as a platform on which to explore new algorithms and architectures for basecalling. The current version uses a bidirectional RNN with LSTM cells and CTC loss to call bases from raw signal, and has been inspired by other community basecallers such as DeepNano and Chiron.