









□ Riemannian Stein Variational Gradient Descent for Bayesian Inference
>> https://arxiv.org/abs/1711.11216
Riemannian Stein Variational Gradient Descent (RSVGD), which has many non-trivial considerations beyond SVGD and requires novel treatments. derive the Riemannian counterpart of the directional derivative, then conceive a novel technique to find the functional gradient, in which case the SVGD technique fails. Finally express RSVGD in the embedded space of the manifold and give an instance for hyperspheres, which is directly used for its application.

□ Aging in a relativistic biological space-time:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/05/229161.full.pdf
extend the concepts of physical space and time to an abstract, mathematically-defined space as the Cartesian product of manifolds and sub-manifolds, which they associate with a concept of “biological space-time” in which biological clocks operate.
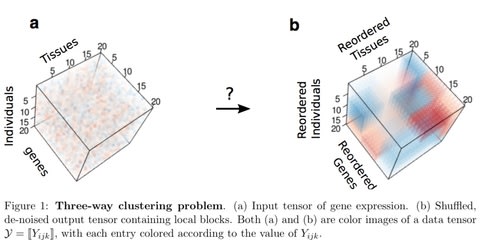
□ Three-way clustering of multi-tissue multi-individual gene expression data using constrained tensor decomposition:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/05/229245.full.pdf
applied MultiCluster to identify 3-way blocks in noisy expression tensors simulated from three different cluster models: additive-, multiplicative-, and combinatorial-mean models. Through simulation and application to the GTEx RNA-seq data, this tensor decomposition identifies three-way clusters with higher accuracy, while being 11x faster, than the competing Bayesian method.
Both MultiCluster and SDA are built upon the Canonical Polyadic (CP) decomposition which decompose a tensor into a sum of rank-1 matrices, whereas HOSVD decomposes a tensor into a core tensor multiplied by an orthogonal matrix in each mode.

□ DeepVariant: Highly Accurate Genomes With Deep Neural Networks:
>> https://research.googleblog.com/2017/12/deepvariant-highly-accurate-genomes.html
The cost-optimized version of DeepVariant calls an aligned 30x genome in 3-4 hours for approximately $8-9 in cloud costs, and an aligned exome in a little over an hour for approximately $0.70.
PROJECT_ID=[your alphanumeric project ID]
OUTPUT_BUCKET=gs://OUTPUT_BUCKET
STAGING_FOLDER_NAME=[a unique alphanumeric name for each run]
OUTPUT_FILE_NAME=output.vcf
MODEL=gs://deepvariant/models/DeepVariant/0.4.0/DeepVariant-inception_v3-0.4.0+http://cl-174375304.data -wgs_standard

□ Dynamical compensation and structural identifiability of biological models: Analysis, implications, and reconciliation:
>> http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005878
DC-Id captures the biological meaning of the dynamical compensation phenomenon, which is the invariance of the dynamics of certain state variables of interest with respect to changes in the values of certain parameters. STRIKE-GOLDD (STRuctural Identifiability taKen as Extended-Generalized Observability with Lie Derivatives and Decomposition) is a methodology and a tool for structural identifiability analysis which can handle nonlinear systems of a very general class, incl. non-rational ones.
□ BioModels performance boost, new model formats
>> https://www.ebi.ac.uk/about/news/announcements/biomodels-performance-boost-new-model-formats
BioModels now supports models built using a wider variety of modelling software and formats and standards, including Python, Mathematica, Matlab SimBiology, etc., as well as SBML and CellML.
□ apwiita:
Fascinating, shedding light on the Dark Proteome: existing genes may harbor additional translation start sites leading to thousands of short "alternative proteins"
□ Deep transcriptome annotation enables the discovery and functional characterization of cryptic small proteins.
>> https://elifesciences.org/articles/27860
alternative proteins with signal peptides (SP) and/or transmembrane domains (TM) predicted by at least two of the three SignalP, PHOBIUS, TMHMM tools and alternative proteins with other signatures. The GO terms assigned to alternative proteins with InterPro entries were grouped and categorized into 13 classes within the three ontologies (cellular component, biological process, molecular function) using the CateGOrizer tool.
□ SpISO-seq: Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome.
>> http://genome.cshlp.org/content/early/2017/12/01/gr.230516.117.abstract
sparse isoform sequencing (spISO-seq), sequences 100k-200k partitions of 10-200 molecules at a time, enabling analysis of 10-100 million RNA molecules. SpISO-seq requires less than 1ng of input cDNA, limiting or removing the need for prior amplification w/ its associated biases.
□ hagentilgner:
Our linked read isoform sequencing approach is out: Lots of coordination of distant splicing events (defined by separate chemical reactions) - you can only see this with long-reads.
#CornellRNAbiology2017 @10xgenomics #transcriptomics #genomics
□ Reevaluation of SNP heritability in complex human traits
>> https://www.nature.com/articles/ng.3865?
empirically derive a model that more accurately describes how heritability varies with minor allele frequency (MAF), linkage disequilibrium (LD) and genotype certainty.
□ HOME: A histogram based machine learning approach for effective identification of differentially methylated regions:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/02/228221.full.pdf
a Histogram Of MEthylation (HOME) based method that exploits the inherent difference in distribution of methylation levels between DMRs and non-DMRs to robustly discriminate between the two via a linear Support Vector Machine. HOME is a highly effective and robust DMR finder that accounts for uneven cytosine coverage in WGBS data, predicts DMRs in various genomic contexts, and accurately identifies DMRs among any number of treatment groups in experiments with or without replicates.
□ GrandPrix: Scaling up the Bayesian GPLVM for single-cell data:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/03/227843.full.pdf
this model is motivated by the DeLorean approach and uses cell capture time to specify a prior over the pseudotime. extending the model to higher-dimensional latent spaces that can be used to simultaneously infer pseudotime and other structure such as branching.
□ Qvella raises US$20M in Series B Financing - new strategic investor bioMérieux will explore collaboration around FAST technology
>> https://docs.wixstatic.com/ugd/45397b_87222b92a3244d1dae3c70bb8d3695ec.pdf
□ Clusterdv, a simple density-based clustering method that is robust, general and automatic.:
>> https://www.biorxiv.org/content/biorxiv/early/2017/11/25/224840.full.pdf
All density-based clustering methods suffer from the problem that density estimation for data with finite sample size produces “sporadic” local maxima that are not related to the “real” structure present in data. As cluster centres with decreasing separability index are added to the dendrogram, they are connected to the cluster centre with which they co-partition at the next higher level.
□ DEAP: Distributed Evolutionary Algorithms in Python
>> https://github.com/DEAP/deap
DEAP is a novel evolutionary computation framework for rapid prototyping and testing of ideas. It seeks to make algorithms explicit and data structures transparent. It works in perfect harmony with parallelisation mechanism such as multiprocessing and SCOOP.
An ephemeral constant is a terminal encapsulating a value that is generated from a given function at run time. Ephemeral constants allow to have terminals that don’t have all the same values.
pset.addEphemeralConstant(lambda: random.uniform(-1, 1))

□ A deep learning method for lincRNA detection using auto-encoder algorithm:
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1922-3
a knowledge-based discovery method using the emerging deep learning technology for lincRNA detection is proposed and developed on DNA genome analysis. It takes advantage of the latest findings of lincRNA data set and aims to utilize the cutting-edge knowledge-based method, namely auto-encoder algorithm, in order to extract the features of lincRNA transcription sites in a more accurate way than conventional methods.
□ pysster: Learning Sequence and Structure Motifs in DNA and RNA Sequences using Convolutional Neural Networks:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/06/230086.full.pdf
The package can be applied to both DNA & RNA to classify sets of sequences by learning sequence & secondary structure motifs. It offers an automated hyper-parameter optimization & options to visualize learned motifs along w/ information about their positional & class enrichment. The Grid_Search class provides a simple way to execute a hyperparameter tuning for the convolutional neural network model. The tuning returns the best model (highest ROC-AUC on the validation data) and an overview of all trained models.
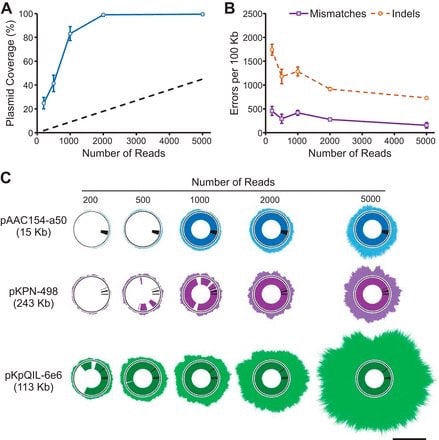
□ Rapid Nanopore Sequencing of Plasmids and Resistance Gene Detection in Clinical Isolates. "...full annotation of plasmid resistance gene content could be obtained in under 6 h from a subcultured isolate"
>> http://jcm.asm.org/content/55/12/3530.abstract
□ Long-Read Sequencing 2017 December 6-7, 2017; BMC Uppsala #LRUA2017
>> https://ngiseminars.wixsite.com/longread2017
the long-read sequencing has revolutionized the field of de novo sequencing for biodiversity studies, making working with this application less time consuming and more productive.
□ Identify causal variants and estimate their effects on splicing in a Massively Parallel Splicing Assay (MaPSy)
>> https://genomeinterpretation.org/content/MaPSy
□ Gene Conversion Facilitates Adaptive Evolution on Rugged Fitness Landscapes
>> http://www.genetics.org/content/207/4/1577
"Our results reveal the potential for duplicate genes to act as a 'scratch paper' that frees evolution from being limited to strictly beneficial mutations in strongly selective environments"

□ VARIMERGE: Succinct De Bruijn Graph Construction for Massive Populations Through Space-Efficient Merging:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/06/229641.full.pdf
reconstruct all edge label positions ephemerally (one base position at a time) in decreasing precedence order, and incrementally refine a data structure that captures the inferable rank and equivalence of full labels based on the portion of the labels this algorithm has seen. two de Bruijn graphs G1 = (V1, E2) and G2 = (V2, E2) constructed with integral value k, without loss of generality, |E1| ≥ |E2|, it follows that VariMerge constructs the merged de Bruijn graph GM in O(m·max(k,t))-time, where t is the number of colors (columns) inCM and m=|E1|.

□ Linked-Read sequencing resolves complex structural variants:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/08/231662.full.pdf
performed 30x Linked-Read genome sequencing on a set of 23 samples with known balanced or unbalanced SVs. Twenty-seven of the 29 known events were detected and another event was called as a candidate. Copy-number variants can be called with as little as 1-2x sequencing depth (5-10Gb) while balanced events require on the order of 10x coverage for variant calls to be made, although specific signal is clearly present at 1-2x sequencing depth.
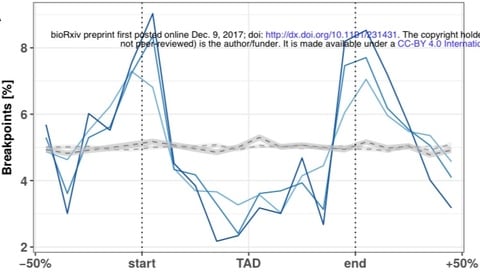
□ Evolutionary stability of topologically associating domains is associated with conserved gene regulation:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/09/231431.full.pdf
Disruptions of TADs by large-scale rearrangements change expression patterns of orthologs across tissues and these changes might be explained by the altered regulatory environment which genes are exposed to after rearrangement. a significant association of conserved GE in TADs & divergent expression patterns in rearranged TADs explaining both why there could be selective pressure on the integrity of TADs over large evolutionary time scales, but also how TAD rearrangement can explain evolutionary leaps.
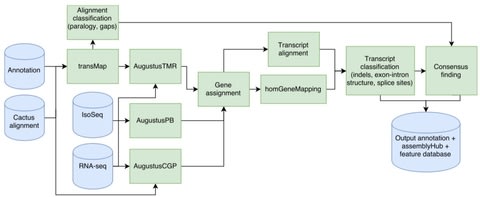
□ Comparative Annotation Toolkit (CAT) - simultaneous clade and personal genome annotation:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/08/231118.full.pdf
The CAT pipeline takes as input a HAL alignment file, an existing annotation set and aligned RNA-seq reads. CAT uses the Cactus alignment to project annotations to other genomes using transMap. AugustusTMR, treats each transcript projection separately and fixes errors in projection. AugustusPB, uses long-read RNA-seq to look for novel isoforms. AugustusCGP uses the Cactus alignment to simultaneously predict protein-coding genes in all aligned genomes.

□ Prometheus: omics portals for interkingdom comparative genomic analyses:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/11/232298.full.pdf
>> http://prometheus.kobic.re.kr
Prometheus supports interkingdom comparative analyses via a domain architecture-based gene identification system, Gene Search, and users can easily and rapidly identify single or entire gene sets in specific pathways. Bioinformatics tools for further analyses are provided in Prometheus or through BioExpress, a cloud-based bioinformatics analysis platform. Prometheus suggests a new paradigm for comparative analyses with large amounts of genomic information.

□ Cell-specific prediction and application of drug-induced gene expression profiles:
>> http://www.worldscientific.com/doi/pdf/10.1142/9789813235533_0004
Expression profiles are compiled into a tensor of 978 genes x 2,130 drugs x 71 cell types. The FaLRTC algorithm is sometimes referred to herein as ‘Tensor’. A three-dimensional tensor can be reshaped or unfolded into matrices in three mathematically distinct ways. The second category of extensions are methodological, including: 1) nonlinear modeling; 2) use of auxiliary similarity information; 3) addition of a time dimension to the tensor; 4) modeling measurement reliability; and 5) adopting a probabilistic framework.
□ LINCSProgram:
A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles:
>> https://www.ncbi.nlm.nih.gov/pubmed/29195078

□ Hercules: a profile HMM-based hybrid error correction algorithm for long reads:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/13/233080.full.pdf
Hercules, the 1st machine learning-based long read error correction algorithm, learns a posterior transition/emission probability distribution for each long read and uses this to correct errors. Hercules decodes the most probable sequence for profile HMM using Viterbi algorithm.
int backtraceWithViterbi(SequencingNode* graph, HMMParameters parameters, double** transitionProbs,
std::pair
std::vector

□ A TOOLBOX TO IMPROVE GENOME ANNOTATION
>> https://www.sib.swiss/about-us/news/1113-a-toolbox-to-improve-genome-annotation
a strategy toward accurate & complete genome annotation consolidates CDSs from multiple reference annotation, ab initio gene prediction algorithms & in silico ORFs (a six-frame translation considering alternative start codons) in an integrated proteogenomics database (iPtgxDB).
□ CLAN: the CrossLinked reads ANalysis tool:
>> https://www.biorxiv.org/content/biorxiv/early/2017/12/14/233841.full.pdf
the existing free energy model and sequence-based interaction model remain insufficient to characterize the complex molecular dynamics, and the computationally predicted RNA secondary structure and RRI remain imprecise. CLAN adopts a dynamic programming-based chaining algorithm to select the two non-overlapping mappings whose total length is maximized.