(Created with Midjourney v6.1)
□ Aaron Hibell / “i feel lost (orchestral reprise)”

□ GENOT: Entropic (Gromov) Wasserstein Flow Matching with Applications to Single-Cell Genomics
>> https://arxiv.org/abs/2310.09254
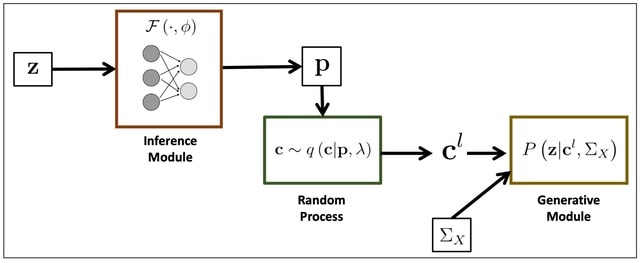
GENOT (Generative Entropic Neural Optimal Transport) is the first method that parameterizes linear and quadratic Entropy-regularized Optimal Transport (EOT) couplings for any cost by modeling their conditional distributions, using flow matching as a backbone.
U-GENOT employs the first neural OT solver for the Fused Gromov-Wasserstein formulation. GENOT can use the geodesic distance on the data manifold, which can be approximated from the shortest path distance on the k-nn graph induced by the Euclidean distance.

□ Evo: Sequence modeling and design from molecular to genome scale
>> https://www.science.org/doi/10.1126/science.ado9336
Evo is a foundation model that is designed to capture two fundamental aspects of biology: the multimodality of the central dogma and the multiscale nature of evolution. Evo learns both of these representations from the whole-genome sequences of millions of organisms.
Evo uses the StripedHyena architecture to enable modeling of sequences at a single-nucleotide, byte-level resolution. Evo has 7 billion parameters and is trained on OpenGenome, a prokaryotic whole-genome dataset containing ~300 billion tokens.

□ scLong: A Billion-Parameter Foundation Model for Capturing Long-Range Gene Context in Single-Cell Transcriptomics
>> https://www.biorxiv.org/content/10.1101/2024.11.09.622759v2
scLong, a billion-parameter foundation model pretrained on 48 million cells. scLong performs self-attention across the entire set of 28,000 genes in the human genome. This enables the model to capture long-range dependencies between all genes.
scLong takes a cell's gene expression vector as input, generating a representation for each element in the vector. Each element corresponds to a specific gene, with its value indicating the level of gene transcription into RNA at a given moment.
scLong leverages Gene Ontology to extract a representation vector for each gene. For each element in the expression vector - defined by a gene ID and its expression value - scLong combines the gene's representation with its expression representation to represent the element.

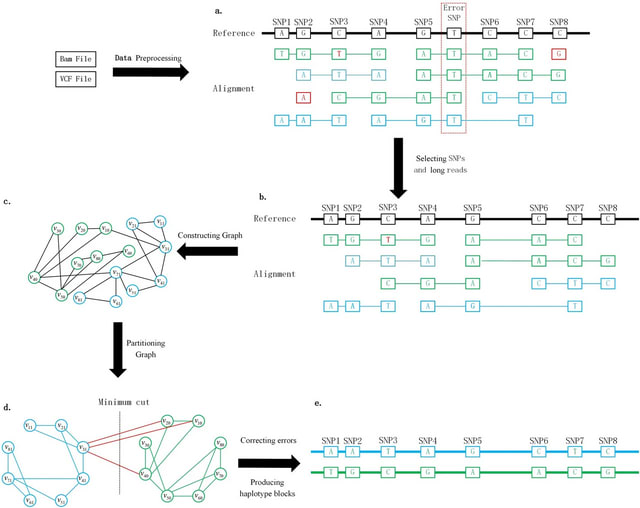
□ devider: long-read reconstruction of many diverse haplotypes
>> https://www.biorxiv.org/content/10.1101/2024.11.05.621838v1
Devider, a new long-read, reference-based haplotyping method for diverse small sequences. Given a set of aligned reads and SNPs, devider models the haplotyping problem as an assembly problem on a positional de Bruijn graph.
devider is inspired by the kSNP algorithm which similarly uses a PDBG but for haplotyping only diploids. The PDBG naturally splits if enough variation is present and collapses under ambiguity, thus haplotyping samples without prior knowledge of the number of distinct sequences.

□ Bio-xLSTM: Generative modeling, representation and in-context learning of biological and chemical sequences
>> https://arxiv.org/abs/2411.04165
Bio-xLSTM introduces three xLSTM-based architectural variants tailored specifically to DNA, proteins and small molecules. They extend XLSTM from causal language modelingto new modeling approaches such as fill-in the middle, in-context learning and masked language modeling.
DNA-xLSTM is an architectural variant tailored for DNA sequences with reverse-complement equivariant blocks, evaluated on long-context generative modeling. DNA-xLSTM-4M has an embedding dimension of 256, 9 mLSTM blocks, and is augmented with Rotary Position Encodings.

□ VI-VS: calibrated identification of feature dependencies in single-cell multiomics
> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03419-z
VI-VS (Variational Inference for Variable Selection) is based on the conditional randomization test (CRT), which quantifies the credibility of pairwise interactions by measuring the effect of exchanging observed features with synthetic ones.
VI-VS harnesses the distributional expressivity of latent variable models. VI-VS relies on deep neural networks for testing, allowing it to scale to large single-cell genomic datasets as well as capture complex nonlinear relationships between variables.

□ REDalign: accurate RNA structural alignment using residual encoder-decoder network
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05956-7
REDalign, a novel method that utilizes the Residual Encoder-Decoder network for RNA structural alignment. In this learning model, the encoder network leverages a hierarchical pyramid to assimilate high-level structural features.
REDalign transforms the pair of input RNA sequences into two-dimensional binary contact matrices in order to represent the relative position of dinucleotides. REDalign learns the conserved structures of RNA sequences and can yield the alignment probability of dinucleotides.
REDalign significantly reduces computational complexity compared to Sankoff-style algorithms and effectively handles non-nested structures, incl. pseudoknots. REDalign can effectively learn residual information and mitigate the vanishing gradient problem.

□ FroM Superstring to Indexing: a space-efficient index for unconstrained k-mer sets using the Masked Burrows-Wheeler Transform (MBWT)
>> https://www.biorxiv.org/content/10.1101/2024.10.30.621029v1
FMSI index, a space-efficient data structure for unconstrained k-mer sets, based on approximated shortest superstrings and the Masked Burrows Wheeler Transform (MBWT), an adaptation of the BWT for masked superstrings.
They prove that 2 + o(1) bits of query memory per indexed k-mer suffice for data sets with spectrum-like property (SLP) and also show that its space requirements grow linearly with the size of the k-mer superstring.
It provides a linear-time construction algorithm and shows how to answer isolated queries in O(k) time and positive streamed queries in O(1) time with an additional bit of memory, with accommodation for reverse complements using saturating counter-based k-mer strand prediction.

□ CelLink: Integrate single-cell multi-omics data with few linked features and imbalanced cell populations
>> https://www.biorxiv.org/content/10.1101/2024.11.08.622745v1
CelLink predicts the cell type for each cell by the weight from the cell-cell transport map. The matched cells will be filtered while the unmatched ones will be re-aligned in the next phase. The transport map will be stored as the cell-cell correspondence matrix.
The second phase is to iteratively align the unmatched cells using unbalanced optimal transport. The iterative alignment is performed separately for each modality. In each iteration, unmatched cells are identified via the cell-cell transport map and re-aligned in the next run.
This transport map is then preserved as the corrected cell-cell correspondence matrix. The alignment stops when the predicted cell type for all unmatched cells does not change, indicating that they cannot be aligned and the model reaches convergence.

□ Multi-context seeds enable fast and high-accuracy read mapping
>> https://www.biorxiv.org/content/10.1101/2024.10.29.620855v1
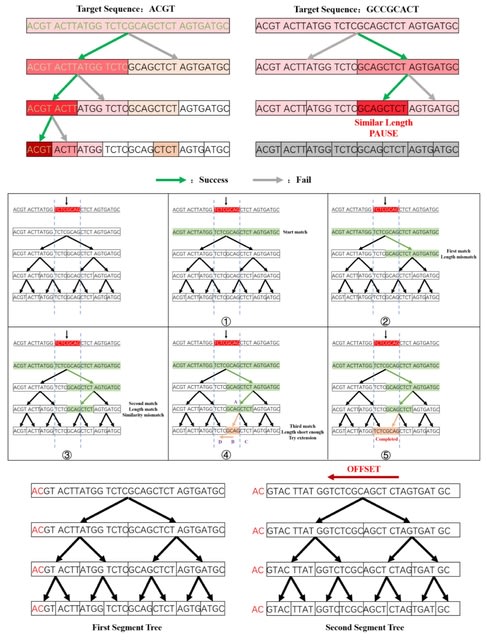
Multi-context seeds (MCS) are strobemers where the hashes of individual strobes are partitioned in the hash value representing the seed. Such partitioning enables a cache-friendly approach to search for both full and partial matches of a subset of strobes.
Strobealign with MCS comes at no cost in memory and only little cost in runtime while offering increased mapping accuracy over default strobealign using simulated Illumina reads across genomes of various complexity.
Strobealign with MCS outperforms minimap2 in short-read mapping and is comparable to BWA-MEM in accuracy in high-variability sequences.

□ noMapper: A mapping-free natural language processing-based technique for sequence search in nanopore long-reads
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05980-7
noMapper is an NLP-based selector designed to search for specific sequences, markers, genes among DNA/RNA long-reads. NoMapper does not use alignment algorithms like Needleman-Wunsch or Smith-Waterman in its work.
All sequences classified as gene/transcript related were subjected to transcript counts per million estimation. noMapper processed 8,000 Nanopore long-reads in about 15 s for dictionary containing 1024 components.

□ llm2geneset: Enhancing Gene Set Overrepresentation Analysis with Large Language Models
>> https://www.biorxiv.org/content/10.1101/2024.11.11.621189v1
llm2geneset, a framework that leverages large language models (LLMs) to dynamically generate gene sets from natural language descriptions. LLM-generated gene sets are significantly overrepresented in corresponding human-curated gene sets.
lIm2geneset generated gene set descriptions had on average a higher fraction of shared unigrams and bigrams as well as higher cosine similarity with the ground truth gene set descriptions than GSAI.

□ GLEANR: Sparse matrix factorization of GWAS summary statistics robust to sample sharing improves detection and interpretation of factors with diverse genetic architectures https://www.biorxiv.org/content/10.1101/2024.11.12.623313v1
GLEANR (GWAS latent embeddings accounting for noise and regularization) uses dynamic model selection to yield sparse factors while statistically accounting for confounding covariance from sample sharing and variation in estimation error.
GLEANR estimates sparse latent genetic components, decomposing the full set of genetic effects across traits into a lower dimensional representation with factors shared by traits.

□ Hyper-k-mers: efficient streaming k-mers representation
>> https://www.biorxiv.org/content/10.1101/2024.11.06.620789v1
Hyper-k-mers, a new k-mer representation that asymptotically decreases duplication. Hyper-k-mers are more succinct than their direct competitor, super-k-mers, achieving space usage closer to sampling techniques such as syncmers, while still providing a direct k-mer representation.
Hyper-k-mers represent sequences as collections of minimizers and the sequences between them. This approach reduces k-mer overlaps and, consequently, theoretically allows hyper-k-mers to achieve a space usage of 4 bits per (DNA) base.
K-mer Fast Counter is a fast and space-efficient k-mer counter based on hyper-k-mers. It is particularly well-suited for counting large k-mers from long reads with a low error-rate. It can filter k-mers based on their count and only retrieve the k-mers above a certain threshold.

□ GROT: Graph-Regularized Optimal Transport for Single-Cell Data Integration
>> https://www.biorxiv.org/content/10.1101/2024.10.30.621072v1
The GROT algorithm begins by constructing a kernel matrix for each single-cell sequencing modality, such as scRNA-Seq and scATAC-Seq. It then learns a mapping matrix to project data from the RKHS space into a shared latent space.
Global alignment within this latent space is facilitated through optimal transport. Graph regularization is applied to maintain the local structural integrity; this assumes that cells close in their original domains should exhibit similar structures in the shared space.

□ GreedyMini: Generating low-density minimizers
>> https://www.biorxiv.org/content/10.1101/2024.10.28.620726v1
GreedyMini, a novel greedy algorithm to generate minimizers with low expected density. At each iteration it chooses uniformly at random one of the k-mers with the lowest score, assigns the next rank to it, and updates the scores of all other unranked k-mers.
GreedyMini+, a novel method to generate low-density DNA minimizers by using the GreedyMini algorithm to generate binary minimizers and transforming them to the DNA alphabet and larger k if needed.

□ col-BWT: Improved pangenomic classification accuracy with chain statistics
>> https://www.biorxiv.org/content/10.1101/2024.10.29.620953v1
col-BWT that enables compressed indexes to generate both matching statistics and chain statistics simultaneously and in linear time with respect to the query length.
Chain statistics complement the fine-grained co-linearity information inherent in MSs and PMLs by additionally conveying whether matches are co-linear with respect to the reference sequences in the index.
col-BWT rapidly compute multi-maximal unique matches (multi-MUMs) and identify BWT sub-runs that correspond to these multi-MUMs.
From these, they select those that can be "tunneled," and mark these with the corresponding multi-MUM identifiers. This yields an O(r + n/d)-space index for a collection of d sequences having a length-n BWT consisting of r maximal equal-character runs.

□ PangeBlocks: customized construction of pangenome graphs via maximal blocks
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05958-5
By leveraging the notion of maximal block in a Multiple Sequence Alignment (MSA), they reframe the pangenome graph construction problem as an exact cover problem on blocks called Minimum Weighted Block Cover.
Each block becomes a vertex of the pangenome graph and we add arcs to the graph to connect blocks that are adjacent in the MSA. An Integer Linear Programming (ILP) formulation for the MWBC problem that allows us to study the most natural objective functions for building a graph.
□ TDFPS-Designer: an efficient toolkit for barcode design and selection in nanopore sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03423-3
TDFPS-Designer, a Designer for a barcode kit that employs a well-defined Threshold to reduce the sampling space of the dynamic time warping (DTW)-based Farthest Point Sampling algorithm for accurate barcoded sample demultiplexing in nanopore sequencing.
TDFPS-Designer selects barcodes within a given sequence space by the farthest point sampling algorithm, directly based on the comparison of nanopore signals.
A DTW distance-based demultiplexing strategy is designed to ensure accurate sample label assignment. Three barcode kits with different barcode lengths were designed by TDFPS-Designer.

□ Blended Length Genome Sequencing (blend-seq): Combining Short Reads with Low-Coverage Long Reads to Maximize Variant Discovery
>> https://www.biorxiv.org/content/10.1101/2024.11.01.621515v1
Blended Length Genome Sequencing (blend-seq), a novel means of combining traditional short-read pipelines with very low coverage long reads. Blend-seq is flexible with respect to choices and coverage levels of each sequencing technology.
Long reads help w/ SNP discovery by better mapping to difficult regions, and they provide better performance with long insertions and deletions by virtue of their length, while the larger number of short-read layers help w/ genotyping structural variants discovered by long reads.

□ isONclust3: De novo clustering of extensive long-read transcriptome datasets
>> https://www.biorxiv.org/content/10.1101/2024.10.29.620862v1
isONclust3 is a greedy algorithm and uses a minimizer-based approach to estimate similarity and to represent clusters. However, isONclust3 addresses the accuracy, time, and memory limitations with isONclust and other algorithms.
The dynamic updating with high-confidence minimizers enables isONclust3 to cluster more reads from the same gene family, while keeping the number of minimizers stored for each cluster informative in comparison to adding all minimizers of a read.

□ The open-closed mod-minimizer algorithm
>> https://www.biorxiv.org/content/10.1101/2024.11.02.621600v1
The open-closed mod-minimizer - a sampling algorithm that has lower density than the best known schemes for k > w, like the mod-minimizer, and also generally works for any value of k.
The open-closed minimizer prefers sampling the smallest open syncmer. If no open syncmer is found in the window, then the smallest closed syncmer is considered, like in miniception. Lastly, if no closed syncmer is found either, the smallest k-mer is sampled.
The open-closed mod-minimizer employs a recursive method to compute the probability distribution of the configurations (O,C, Oc, Cc) and, hence, the density of open-closed minimizers, that runs in time polynomial in the number of s-mers in a context, w + k -s + 1.

□ Vcfexpress: flexible, rapid user-expressions to filter and format VCFs
>> https://www.biorxiv.org/content/10.1101/2024.11.05.622129v1
Vefexpress is implemented in the rust programming language using rust-htslib which wraps the HTSlib C library.
Vcfexpress is nearly as fast as BCFTools, but adds functionality to execute user expressions in the lua programming language for precise filtering and reporting of variants from a VCF or BCF file.

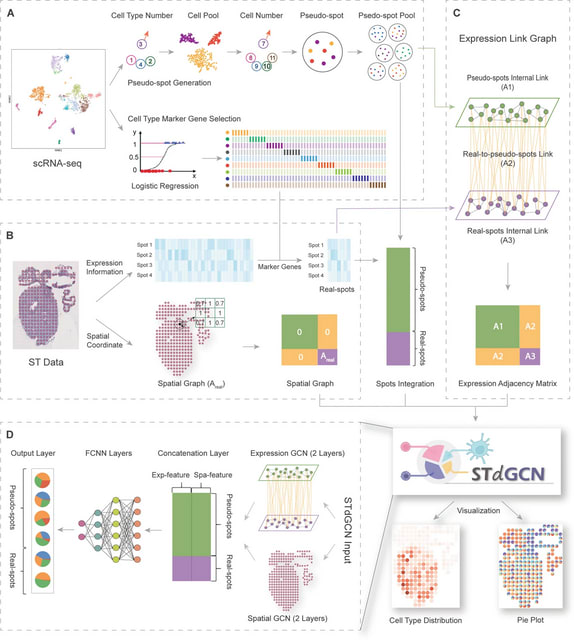
□ GraphPCA: a fast and interpretable dimension reduction algorithm for spatial transcriptomics data
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03429-x
GraphPCA, a quasi-linear dimension-reduction algorithm tailored for ST data. GraphPCA learns the low-dimensional representation of ST data based on PCA with minimum reconstruction error, by incorporating spatial location information as constraints in the reconstruction step.
GraphPCA infers an embedding matrix integrating both spatial location and gene expression information by solving an optimization problem with constraints determined by the constructed spatial neighborhood graph.

□ mcBERT: Patient-Level Single-cell Transcriptomics Data Representation
>> https://www.biorxiv.org/cgi/content/short/2024.11.04.621897v1
mcBERT(multi-cell BERT) integrates a transformer encoder and multiple data sources, mcBERT processes individual cell gene counts to produce a compact, disease-capturing patient vector that condenses relevant information learned from single-cell gene expressions.
mcBERT uses a self-supervised data2vec methodology. Post-embedding, all cellular data is averaged to generate a singular patient-level vector, which is utilized during inference with the refined model weights.
mcBERT begins with a non-contextual cell embedding via a linear layer, followed by a transformer encoder composed of 12 blocks, each with 12 attention heads, to contextualize the cell data. It has a hidden dimensionality of 288.

□ scDOT: optimal transport for mapping senescent cells in spatial transcriptomics
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03426-0
scDOT employs a probabilistic mapping, where the precision of the mapping is modulated by incorporating the coarse-grained mapping of cell types obtained from the deconvolution task. scDOT uses a bilevel optimization approach, based on the differentiable deep declarative network.
scDOT simultaneously and in parallel learns the cell type fraction of each spot (deconvolution task) and the mapping between individual cells in the scRNA-seq data and individual spots in the spatial transcriptomics data (spatial reconstruction task).
The resulting mapping matrix between cells and spots is then utilized to construct the cell-cell spatial neighborhood graph, where cells are connected if they are in close physical proximity.

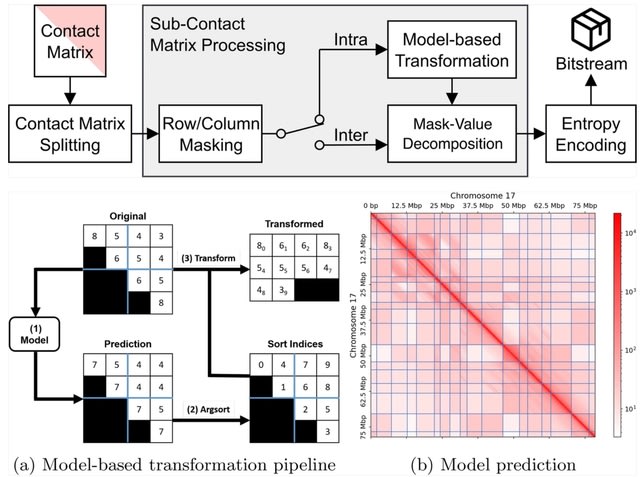
□ scHiCcompare: an R package for differential analysis of single-cell Hi-C data
>> https://www.biorxiv.org/content/10.1101/2024.11.06.622369v1
scHiCcompare imputes single-cell matrices while maintaining both genomic distance effects and cell-specific variability, employing a pseudo-bulk strategy to normalize pseudo-bulk matrices from two groups of scHi-C data.
scHiCcompare demonstrates robust performance across various genomic distance ranges. Random forest models with progressive and Fibonacci pooling produced distributions closest to the observed IF distribution with targeted genomic distances.

□ BaNDyT: Bayesian Network modeling of molecular Dynamics Trajectories
>> https://www.biorxiv.org/content/10.1101/2024.11.06.622318v1
BaNDyT is the first software package to include specialized and advanced features for analyzing MD simulation trajectories using a probabilistic graphical network model.
BaNDyT utilizes a maximum entropy binning algorithm for discretization. This approach ensures that each bin contains a roughly equal number of data points, while also maintaining as much randomness as possible in the allocation of data to bins.

□ easySHARE-seq: Flexible and high-throughput simultaneous profiling of gene expression and chromatin accessibility in single cells
>> https://www.biorxiv.org/content/10.1101/2024.02.26.581705v3
easySHARE-seq, an elaboration of SHARE-seq, for the simultaneous measurement of ATAC- and RNA-seq in single cells.
easySHARE-seq generates accurate and reproducible data, that both modalities can be used to identify the same cell types and how it compares to other technologies in terms of data quality. easySHARE-seq leverages the simultaneous measurements to identify peak-gene interactions.

□ scSimu: Inferring Cell-Type-Specific Co-Expressed Genes from Single Cell Data
>> https://www.biorxiv.org/content/10.1101/2024.11.08.622700v1
scSimu (Single-Cell SIMUlation) improves the simulation method to generate scRNA-seq count data from real data. scSimu is specifically developed for evaluating gene co-expression networks estimated from Drop-seq scRNA-seq data.
The simulation model accommodates discrete covariates, enabling the generation of data for each distinct category which can then be concatenated. It also preserves all gene names and does not eliminate any genes or cells.

□ MAGE: Monte Carlo method for Aberrant Gene Expression
>> https://www.biorxiv.org/content/10.1101/2024.11.08.622686v1
MAGE can identify aberrantly expressed genes (AEGs) that are not found by conventional DE analyses. MAGE can identify outliers based on the expression profile of all genes rather than performing DE analyses on a per-gene basis.

□ NOODAI: A webserver for network-oriented multi-omics data analysis and integration pipeline
>> https://www.biorxiv.org/content/10.1101/2024.11.08.622488v1
NOODAI, an online platform for the combined analysis of multiple omics profiles. The tool takes as input user-provided lists of hits for different analyzed omics layers and maps them onto a high-confidence molecular interaction network.
NOODAl maps the provided elements onto biological networks, identifies elements that are central for network connectivity and reports network modules with highly connected elements.

□ scExplorer: A Comprehensive Web Server for Single-Cell RNA Sequencing Data Analysis
>> https://www.biorxiv.org/content/10.1101/2024.11.11.622710v1
In scExplorer, users can select between the Seurat and Cell Ranger methods for HVG identification. scExplorer enables users to conduct doublet detection with Scrublet, batch correction using several tools and flexible export options in python and R formats.
The method implemented in Seurat stabilizes variance across datasets, particularly in multi-batch analyses, while the approach available from Cell Ranger ensures optimal performance for datasets generated using 10x Genomics pipelines.























































































































































































































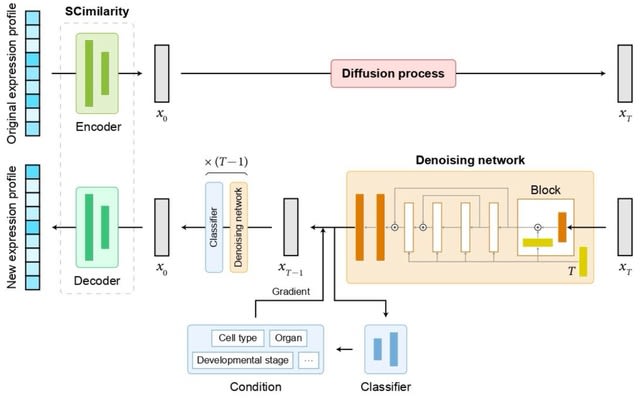
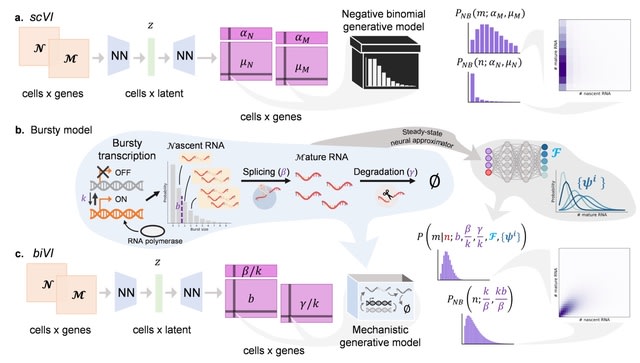
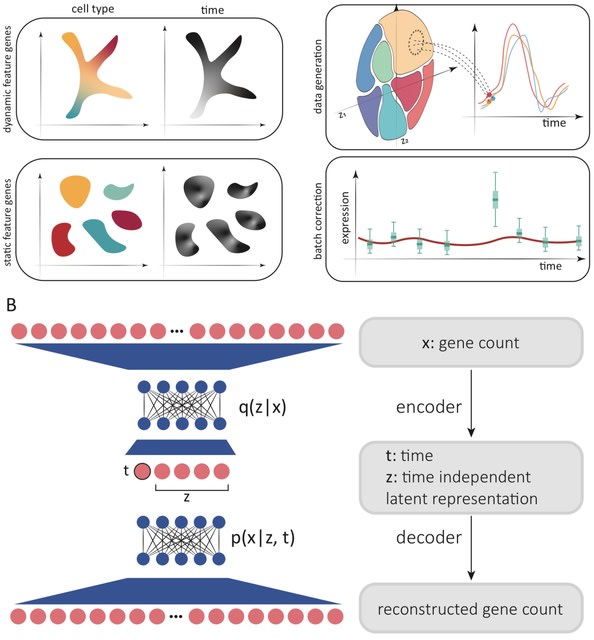

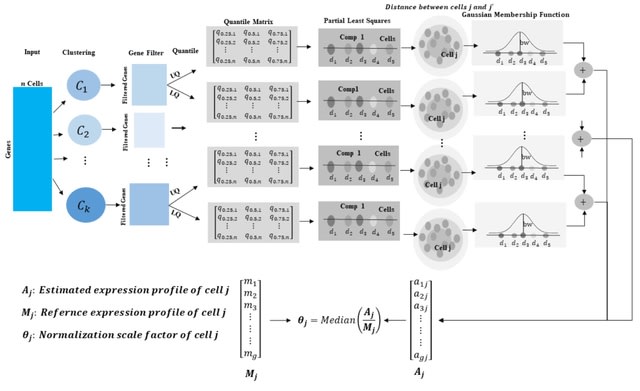
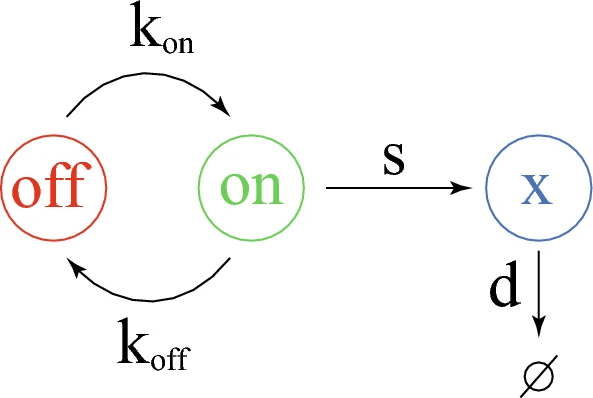
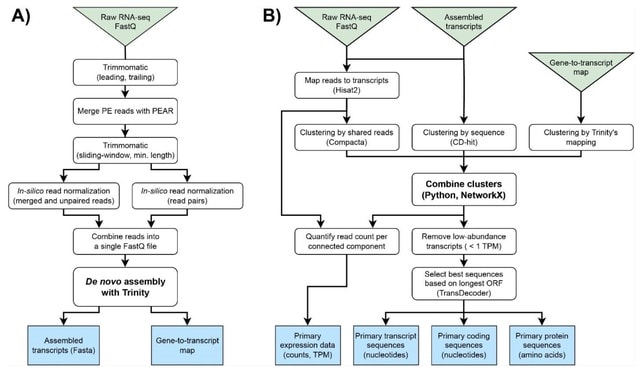
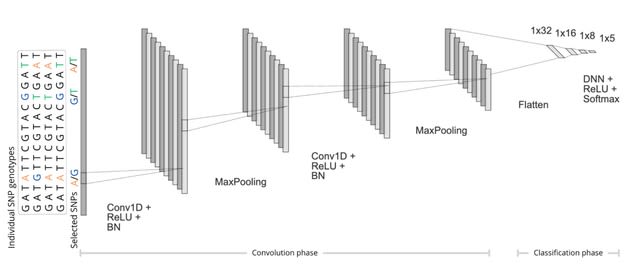

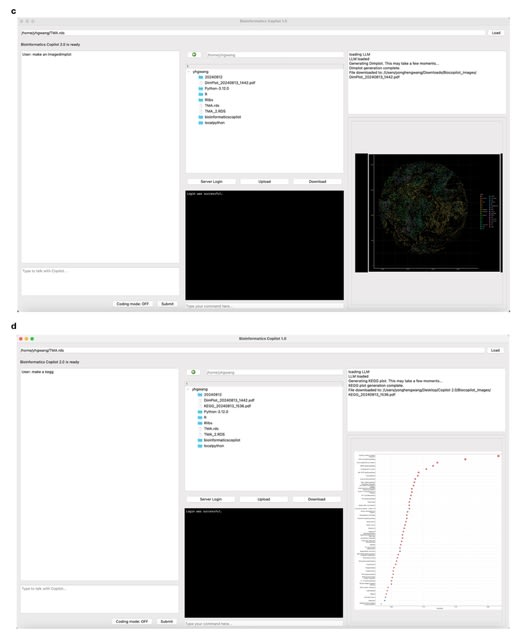
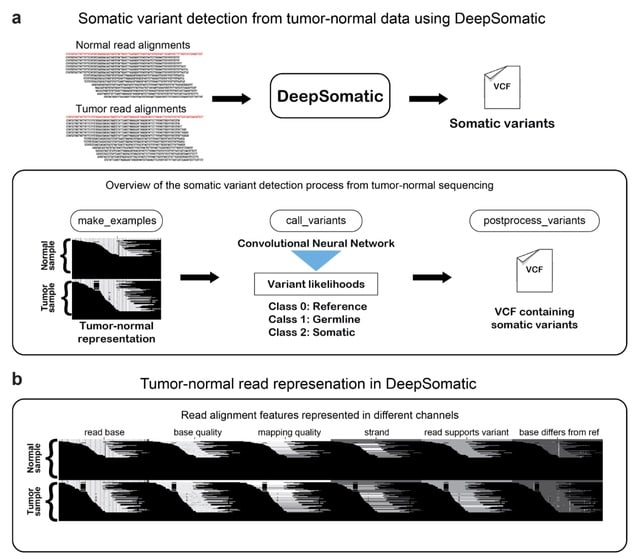
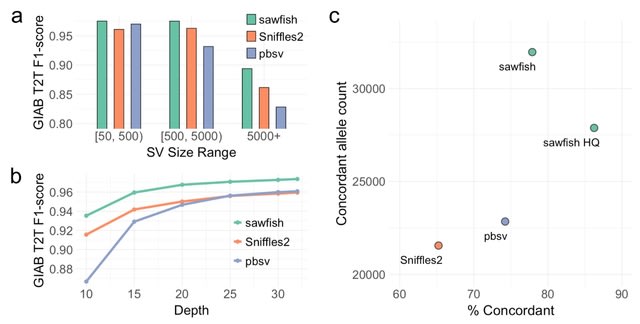
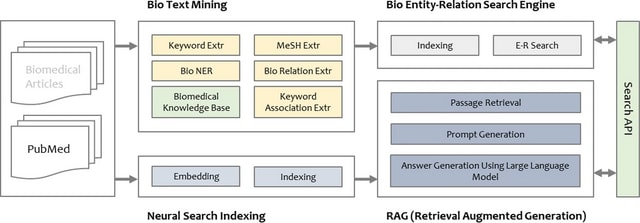
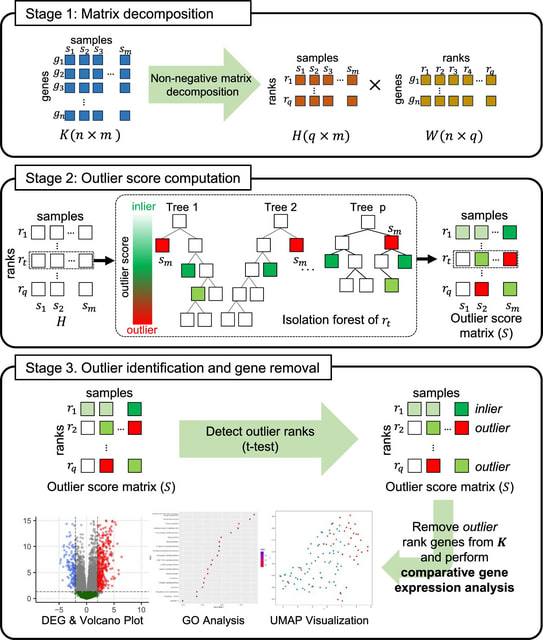

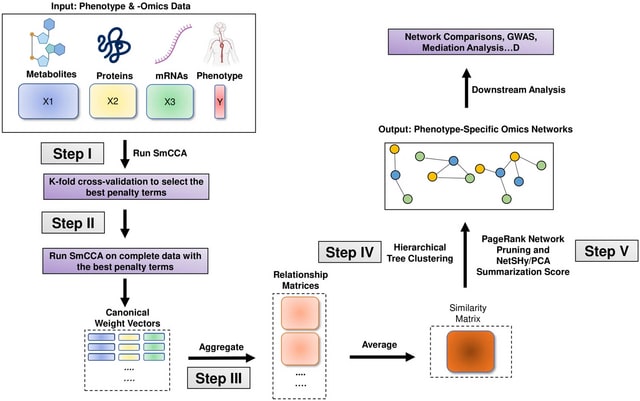







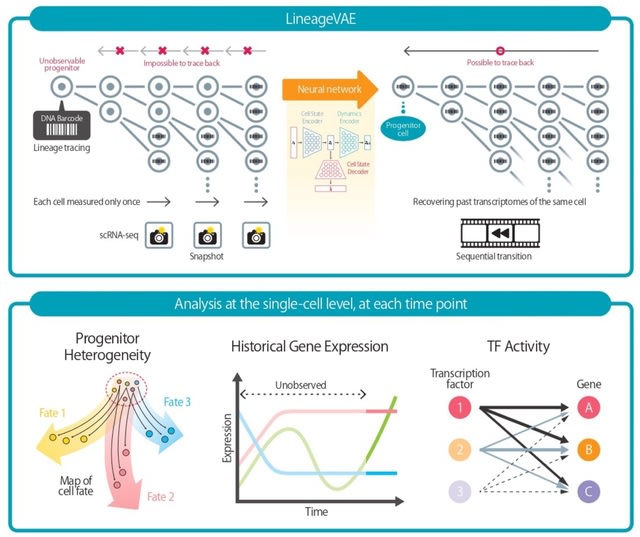









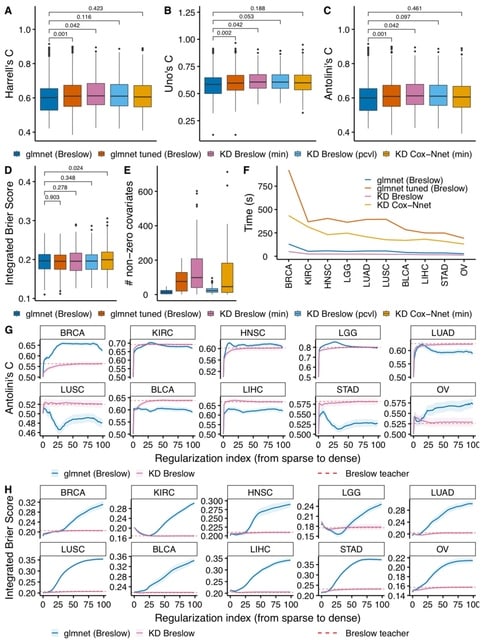





























 □ DeepCSCN: Deep Learning Driven Cell-Type-Specific Embedding for Inference of Single-Cell Co-expression Networks
□ DeepCSCN: Deep Learning Driven Cell-Type-Specific Embedding for Inference of Single-Cell Co-expression Networks