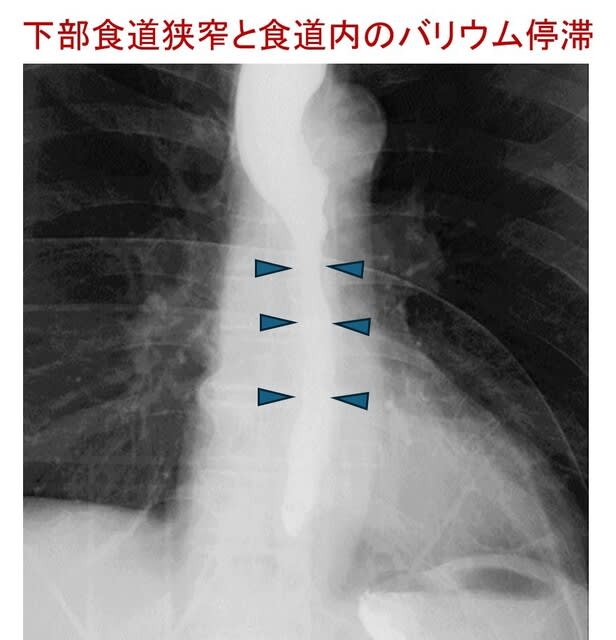
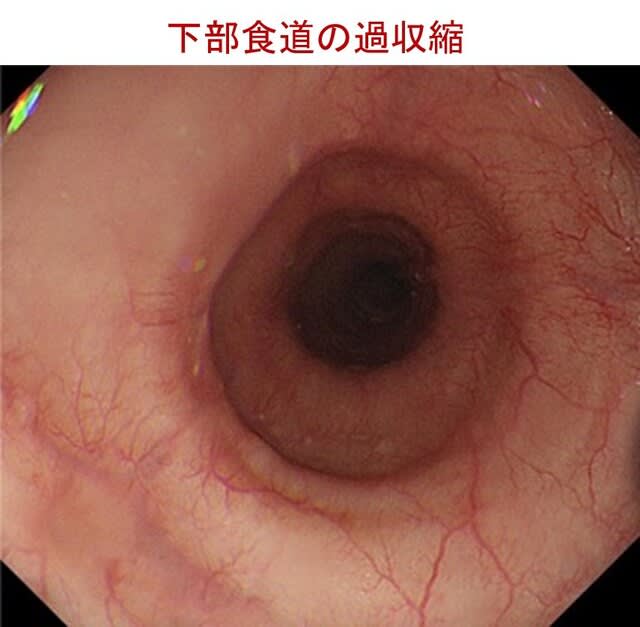
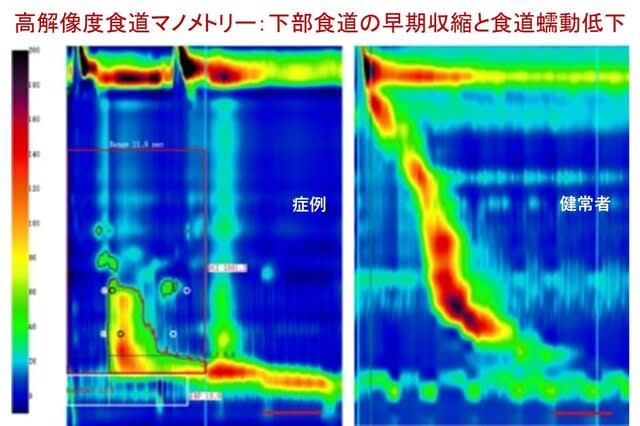
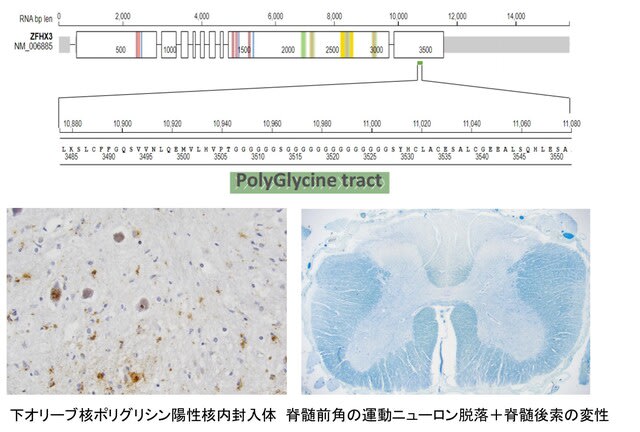
当科の森泰子先生らは,失調性歩行障害を主訴とする80歳男性を担当しました.家族歴なし.73歳より月1-2回,短期間,手を握ると開きにくくなることを自覚するようになり,また一過性の構音障害も経験しました.一過性脳虚血発作が疑われたものの各種検査で異常はなく,その後,構音障害と歩行困難は徐々に悪化し持続性になりました.慢性咳嗽なし.82歳時の神経症候では,小脳性運動失調,手袋・靴下型感覚障害,両側前庭動眼反射の障害がみられ,眼振はなし.手指には萎縮と拘縮がみられました.手指の曲げ伸ばしはでき,強く握ると開きにくい症状は残存していました(把握ミオトニア:図).叩打ミオトニアなし.RFC1遺伝子,DMPK遺伝子とも変異なし.

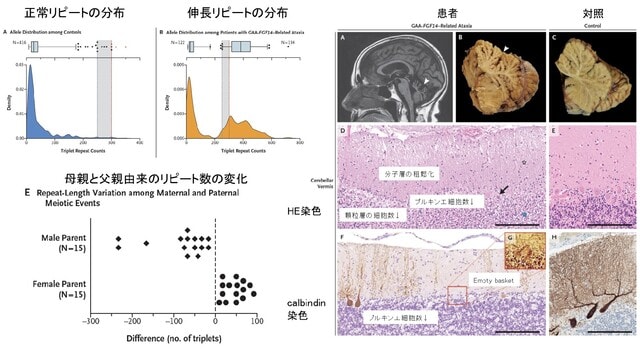
しかしFGF14遺伝子イントロンに229の純粋GAAリピート伸長を認めました.脊髄小脳失調症27 B(SCA27B)の原因遺伝子です.初期の報告では250リピートが病原性の閾値と示唆されていましたが,近年,200〜250のリピートが本症を発症しうることを示唆する報告がなされており,229リピートは病的である可能性があります.本例の特徴は初発症状のエピソード性の把握ミオトニアですが,SCA27Bではエピソード性運動失調を示すこと,またFGF14は神経系において電位依存性ナトリウムチャネルの機能を調節していることから説明ができなくもないように思います.今後の症例の集積が必要と思われます.なお本研究は横浜市立大学宮武聡子先生,輿水江里子先生,松本直通教授との共同研究として行いました.
Mori Y, Miyatake S, Kunieda K, Yoshikura N, Hayashi Y, Higashida K, Kimura A, Koshimizu E, Matsumoto N, Shimohata T. A cerebellar ataxia patient harboring 229 pure GAA repeat units in FGF14 presenting with grip myotonia. Neurol Clin Neurosci. 06 May 2024(doi.org/10.1111/ncn3.12826)
しかしFGF14遺伝子イントロンに229の純粋GAAリピート伸長を認めました.脊髄小脳失調症27 B(SCA27B)の原因遺伝子です.初期の報告では250リピートが病原性の閾値と示唆されていましたが,近年,200〜250のリピートが本症を発症しうることを示唆する報告がなされており,229リピートは病的である可能性があります.本例の特徴は初発症状のエピソード性の把握ミオトニアですが,SCA27Bではエピソード性運動失調を示すこと,またFGF14は神経系において電位依存性ナトリウムチャネルの機能を調節していることから説明ができなくもないように思います.今後の症例の集積が必要と思われます.なお本研究は横浜市立大学宮武聡子先生,輿水江里子先生,松本直通教授との共同研究として行いました.
Mori Y, Miyatake S, Kunieda K, Yoshikura N, Hayashi Y, Higashida K, Kimura A, Koshimizu E, Matsumoto N, Shimohata T. A cerebellar ataxia patient harboring 229 pure GAA repeat units in FGF14 presenting with grip myotonia. Neurol Clin Neurosci. 06 May 2024(doi.org/10.1111/ncn3.12826)