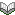免疫チェックポイント阻害剤(Immune Checkpoint Inhibitors;ICI)は,悪性黒色腫や非小細胞肺がんなど多くのがんで顕著な治療効果を示し,がん治療に画期的な進歩をもたらしました.しかし免疫系の「ブレーキ」を解除することによって,自己免疫関連副作用(immune-related adverse events;irAEs)が発生するリスクも伴います.その中で,脳神経内科領域の副作用として頻度が高いのがICI関連筋症(ICI-myopathy)です.私どもの施設は大学病院ということもありますが,最近非常に増加している印象があります.Neurology誌に掲載された総説論文では,ICI関連筋症の発症メカニズムについて詳しく解説しており,勉強になりました.
免疫チェックポイントは,CTLA-4(細胞傷害性Tリンパ球抗原4)やPD-1(プログラム細胞死1),PD-L1(プログラム細胞死リガンド1),LAG-3(リンパ球活性化遺伝子3)を介してT細胞の過剰な活性化を抑える「ブレーキ」として機能しています.しかし,がん細胞はこの免疫抑制機構を利用して免疫系からの攻撃を回避します.ICIはこのブレーキを解除し,T細胞が再びがん細胞を攻撃できるようにする治療法です.しかしICIによってT細胞が活性化されると,がん細胞だけでなく,正常な筋細胞も誤って標的とされることがあります.
図1は,ICI関連筋症のメカニズムを示しています.がん細胞(黒色腫)が死滅すると抗原提示細胞(APC)ががん抗原を取り込み,局所リンパ節でT細胞に抗原を提示します.この過程でCTLA-4,PD-1,PD-L1,LAG-3の働きがICIで阻害されると,T細胞は過剰に活性化され,がん細胞への免疫応答が強化されます.しかし正常組織も攻撃対象となります.特にCD8+ T細胞が活性化しますが,IL-6経路の活性化に伴い,さらに病原性の高いTh17細胞やTc17細胞へと分化させ,これらが自己の筋細胞への攻撃をもたらします(ともに炎症反応や自己免疫疾患の病態において重要な役割を果たすT細胞のサブセット).こうした炎症反応の結果として,筋線維の壊死と再生が繰り返され,臨床的には筋力低下,嚥下障害,呼吸不全等が生じます.

なぜ筋組織が標的となるのかはよく分かっていないうようです.仮説としては,がん抗原と筋細胞の抗原が類似しているため,「分子模倣(molecular mimicry)」現象が生じ,誤って筋細胞が攻撃されるというものがあります.例えば,心筋炎を合併するICI関連筋症の患者では,心筋と骨格筋の両方に共通するα-ミオシンなどに対する免疫反応が確認されています.また筋線維自体が抗原提示分子(MHCクラスI)を発現する能力を持つことが関連しているとも言われています.通常,筋細胞は免疫系の標的となりにくい組織ですが,炎症環境ではMHCクラスIの発現が増加し,自己免疫攻撃を受けやすくなる可能性があるようです.
以下,論文の病態機序以外のメモです.
◆頻度:ICI治療を受ける患者の0.4%〜0.8%で発症する.
◆発症様式: 急性または亜急性の眼筋,球麻痺,近位筋の筋力低下が特徴.1/3の患者で心筋炎を合併し,その場合,死亡率は50%と上昇する.
◆誤診のリスク: 眼筋症状は重症筋無力症と誤診されやすいが,疲労性の筋力低下は見られない.また神経筋接合部障害の電気生理学的所見も認められないことが多い.AChR抗体は最大40%で陽性.
◆画像検査:MRI T2WIで筋の浮腫性変化や造影増強効果あり.FDG-PETで筋組織の代謝亢進を確認可能.
◆筋生検:壊死および再生筋線維の多発性クラスターが特徴.CD4+およびCD8+リンパ球,マクロファージの浸潤.MHC-Iの過剰発現,IL-6経路の活性化.
◆遺伝子解析: インターフェロン経路やIL-6経路の活性化が示唆される.
◆治療としては,従来の高用量ステロイド療法に加えて,IL-6阻害薬(トシリズマブ)やJAK阻害薬の可能性が議論されている.
◆CTCAE(Common Terminology Criteria for Adverse Events)グレードが報告されている.グレードごとに症状の重症度が定義されており,それに応じた治療戦略が推奨されている(図2).
Beecher G, et al. Immune Checkpoint Inhibitor Myopathy: The Double-Edged Sword of Cancer Immunotherapy. Neurology. 2024;103:e210031. (doi.org/10.1212/WNL.0000000000210031)

免疫チェックポイントは,CTLA-4(細胞傷害性Tリンパ球抗原4)やPD-1(プログラム細胞死1),PD-L1(プログラム細胞死リガンド1),LAG-3(リンパ球活性化遺伝子3)を介してT細胞の過剰な活性化を抑える「ブレーキ」として機能しています.しかし,がん細胞はこの免疫抑制機構を利用して免疫系からの攻撃を回避します.ICIはこのブレーキを解除し,T細胞が再びがん細胞を攻撃できるようにする治療法です.しかしICIによってT細胞が活性化されると,がん細胞だけでなく,正常な筋細胞も誤って標的とされることがあります.
図1は,ICI関連筋症のメカニズムを示しています.がん細胞(黒色腫)が死滅すると抗原提示細胞(APC)ががん抗原を取り込み,局所リンパ節でT細胞に抗原を提示します.この過程でCTLA-4,PD-1,PD-L1,LAG-3の働きがICIで阻害されると,T細胞は過剰に活性化され,がん細胞への免疫応答が強化されます.しかし正常組織も攻撃対象となります.特にCD8+ T細胞が活性化しますが,IL-6経路の活性化に伴い,さらに病原性の高いTh17細胞やTc17細胞へと分化させ,これらが自己の筋細胞への攻撃をもたらします(ともに炎症反応や自己免疫疾患の病態において重要な役割を果たすT細胞のサブセット).こうした炎症反応の結果として,筋線維の壊死と再生が繰り返され,臨床的には筋力低下,嚥下障害,呼吸不全等が生じます.
なぜ筋組織が標的となるのかはよく分かっていないうようです.仮説としては,がん抗原と筋細胞の抗原が類似しているため,「分子模倣(molecular mimicry)」現象が生じ,誤って筋細胞が攻撃されるというものがあります.例えば,心筋炎を合併するICI関連筋症の患者では,心筋と骨格筋の両方に共通するα-ミオシンなどに対する免疫反応が確認されています.また筋線維自体が抗原提示分子(MHCクラスI)を発現する能力を持つことが関連しているとも言われています.通常,筋細胞は免疫系の標的となりにくい組織ですが,炎症環境ではMHCクラスIの発現が増加し,自己免疫攻撃を受けやすくなる可能性があるようです.
以下,論文の病態機序以外のメモです.
◆頻度:ICI治療を受ける患者の0.4%〜0.8%で発症する.
◆発症様式: 急性または亜急性の眼筋,球麻痺,近位筋の筋力低下が特徴.1/3の患者で心筋炎を合併し,その場合,死亡率は50%と上昇する.
◆誤診のリスク: 眼筋症状は重症筋無力症と誤診されやすいが,疲労性の筋力低下は見られない.また神経筋接合部障害の電気生理学的所見も認められないことが多い.AChR抗体は最大40%で陽性.
◆画像検査:MRI T2WIで筋の浮腫性変化や造影増強効果あり.FDG-PETで筋組織の代謝亢進を確認可能.
◆筋生検:壊死および再生筋線維の多発性クラスターが特徴.CD4+およびCD8+リンパ球,マクロファージの浸潤.MHC-Iの過剰発現,IL-6経路の活性化.
◆遺伝子解析: インターフェロン経路やIL-6経路の活性化が示唆される.
◆治療としては,従来の高用量ステロイド療法に加えて,IL-6阻害薬(トシリズマブ)やJAK阻害薬の可能性が議論されている.
◆CTCAE(Common Terminology Criteria for Adverse Events)グレードが報告されている.グレードごとに症状の重症度が定義されており,それに応じた治療戦略が推奨されている(図2).
Beecher G, et al. Immune Checkpoint Inhibitor Myopathy: The Double-Edged Sword of Cancer Immunotherapy. Neurology. 2024;103:e210031. (doi.org/10.1212/WNL.0000000000210031)