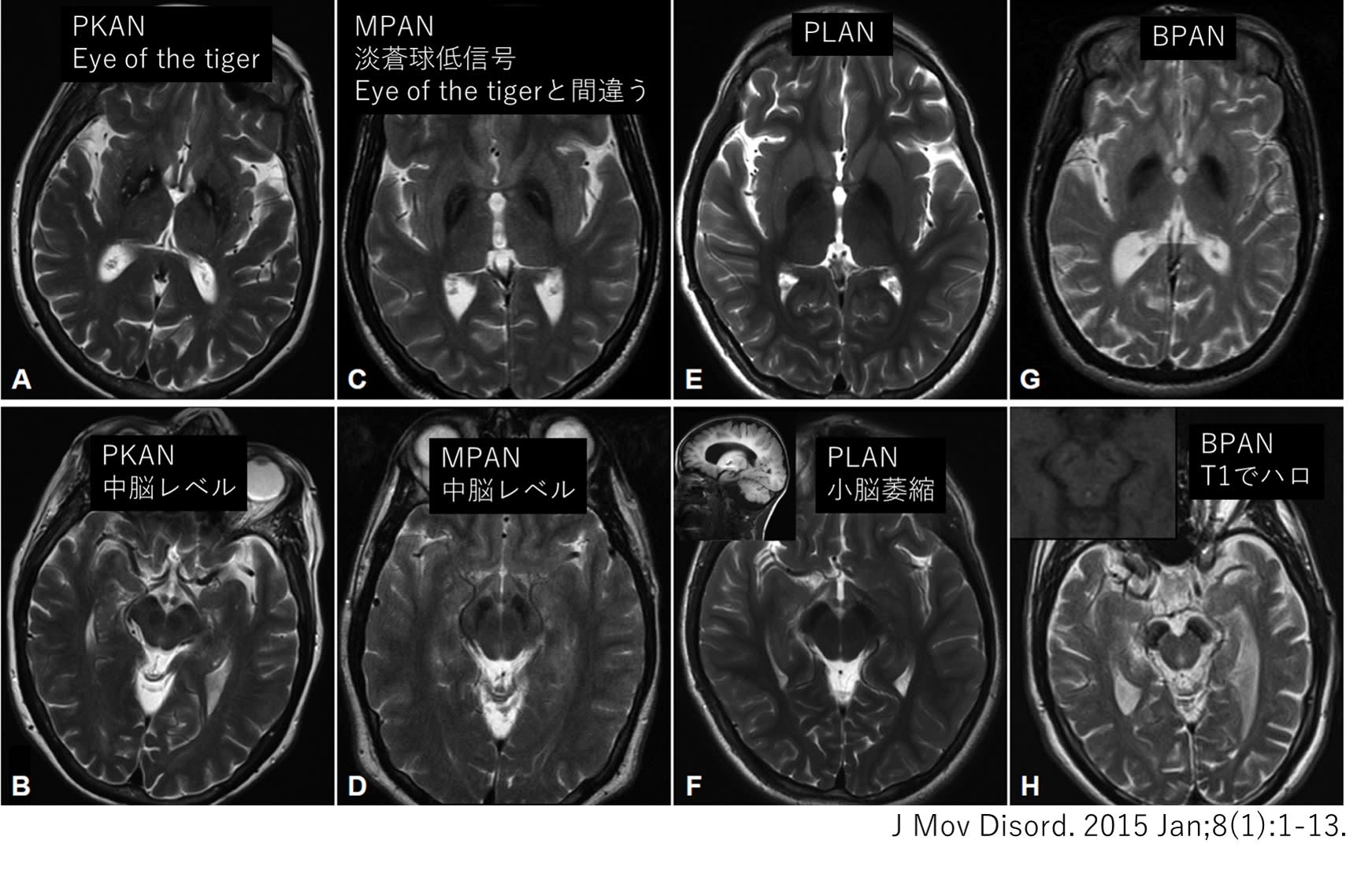
ハンチントン病(HD)は,常染色体顕性遺伝を呈する神経変性疾患で,原因は huntingtin(HTT)遺伝子内のCAGリピートの異常伸長です.HD患者は36回以上(ほとんどの場合40~49回)になります.未解明な点が多く,代表的なものとしては以下が挙げられます.
1)細胞型特異性:主に線条体の中型有棘ニューロン(SPNs)が喪失するが,インターニューロンやグリア細胞は生存すること.
2)長い潜伏期間:典型的には40~50歳で運動症状が現れるが,それ以前は認知機能や運動機能が健常者と同程度であること.
3)HTT遺伝子変異が神経変性を引き起こす具体的なメカニズム
最新号のCell誌に,マサチューセッツ工科大学およびハーバード大学などの研究チームが,体細胞内でのDNAリピート拡大がこの疾患にどのように影響するかを明らかにした研究が報告されました.これまで脳組織全体や血液サンプルから抽出したDNAを用いてリピートの長さを測定する方法が一般的でしたが,この研究では単一細胞でのCAGリピート長の測定と,同一細胞でのRNA発現解析を同時に行える技術を開発して検討した点が画期的と言えます.
研究チームは,患者群50名と対照群53名の線条体組織を解析し,線条体ニューロンの減少が病気の進行と共に顕著になることを示しています.直接経路(線条体から出発し,直接,内節淡蒼球および黒質網様部に投射する)および間接経路(線条体から出発し,まず外節淡蒼球に信号を送り,そこから視床下核を介して淡蒼球内節および黒質網様部に間接的に投射する)という2種類のニューロンについて解析を行い,間接経路ニューロンが早期に失われやすいことが明らかにしました.この変化は舞踏運動の出現に関与している可能性があります.
また患者脳を細胞ごとに解析し,HDを引き起こすHTT遺伝子のCAGリピートが,病気の進行に伴い体細胞内で増加することを発見しました!とくに線条体のニューロンでは,初期にはリピートが安定しているものの,加齢とともに徐々に伸長し,特定の閾値を超えると急速に毒性を発揮することを示しました.この現象は「somatic expansion(体細胞伸長)」と呼ばれ,白血球DNAでは見られない脳特有の変化であることも確認されました.
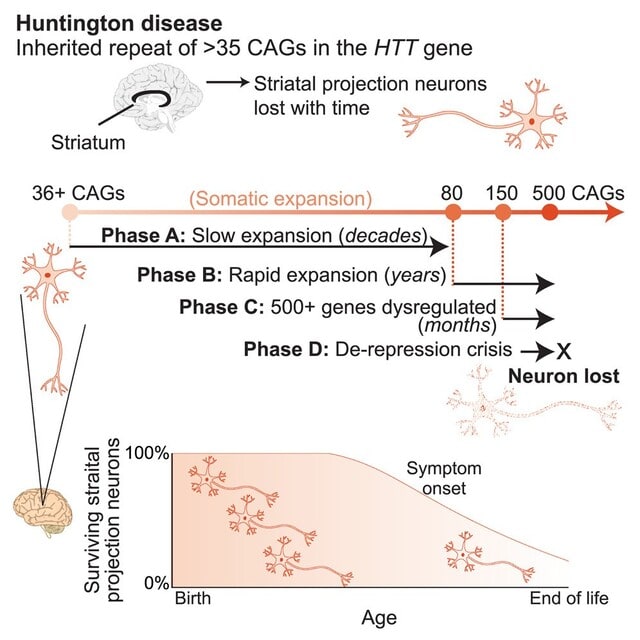
注目すべき発見は,CAGリピートの数が150を超えるとニューロンが急速に傷害されるという点です.CAGリピートは通常は40程度ですが,時間が経過し80を超えると急速に拡大し,150,500を超えると神経毒性を持ちます.つまり「変異したHTTタンパク質の慢性的な毒性」ではなく,「リピート数が閾値を超えることで突然細胞が死ぬ」という新たなメカニズムを提唱するものです.これがニューロンの遺伝子発現を変化させ,細胞死を引き起こすということです.図で説明すると,「Phase A」では,体細胞内でCAGリピートが緩やかに拡張する.この段階は数十年という長い時間をかけて進行し,臨床的な症状は現れません.「Phase B」では,CAGリピートが急速に拡張し,短期間(数年)で150リピートを超える段階に達します.この急速な拡張が,遺伝子発現に影響を及ぼす準備段階となります.「Phase C」では,CAGリピートが500回以上に達し,これが原因で500以上の遺伝子発現が異常を来す段階に入ります.この段階は数か月という短期間で進行し,細胞内の転写機構や神経機能に深刻な影響を及ぼします.最終段階である「Phase D」では,脱抑制危機(de-repression crisis)が発生し,神経細胞が不可逆的に死に至ります.このプロセスにより,HDの特徴的な神経変性が完成します.図の下部は,年齢と線条体ニューロンの生存率の関係を示しています.
また研究では体細胞伸長は,DNA修復酵素(特にMSH3)による減少である可能性が高く,修復過程でCAGリピートが誤って伸長されると考えています.よってMSH3や関連する酵素をターゲットにしてCAGリピートの伸長を抑制することで,症状の発現を遅らせたり進行を抑えたりする治療法が考えられます.すでに発症している患者においても,まだ毒性閾値を超えていないニューロンに作用して病気の進行を遅らせる可能性があります.今回の研究は,HDの発症・進行における新しいメカニズムを示すだけでなく,将来の治療法開発に向けた重要な基盤となる可能性があります.
最初に紹介した3つの問題点について完全解明ではないかもしれませんが,大きな進展が得られました.CAGリピート病は私の学位論文のテーマですが,このようなメカニズムがあるとは夢にも思いませんでした.おそらくHD以外のCAGリピートでも同じことが起きているのではないかと思います.SCA6やCAGリピート以外のリピート病(例:筋強直性ジストロフィーや脆弱X症候群など)はどうなのかも気になります.近いうちにどんどんデータが報告されるのではないかと思います.
Handsaker, Robert E. et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease. Cell, January 16,(doi.org/10.1016/j.cell.2024.11.038)
研究チームへのインタビューです.
https://youtu.be/hd8Uukrocps
1)細胞型特異性:主に線条体の中型有棘ニューロン(SPNs)が喪失するが,インターニューロンやグリア細胞は生存すること.
2)長い潜伏期間:典型的には40~50歳で運動症状が現れるが,それ以前は認知機能や運動機能が健常者と同程度であること.
3)HTT遺伝子変異が神経変性を引き起こす具体的なメカニズム
最新号のCell誌に,マサチューセッツ工科大学およびハーバード大学などの研究チームが,体細胞内でのDNAリピート拡大がこの疾患にどのように影響するかを明らかにした研究が報告されました.これまで脳組織全体や血液サンプルから抽出したDNAを用いてリピートの長さを測定する方法が一般的でしたが,この研究では単一細胞でのCAGリピート長の測定と,同一細胞でのRNA発現解析を同時に行える技術を開発して検討した点が画期的と言えます.
研究チームは,患者群50名と対照群53名の線条体組織を解析し,線条体ニューロンの減少が病気の進行と共に顕著になることを示しています.直接経路(線条体から出発し,直接,内節淡蒼球および黒質網様部に投射する)および間接経路(線条体から出発し,まず外節淡蒼球に信号を送り,そこから視床下核を介して淡蒼球内節および黒質網様部に間接的に投射する)という2種類のニューロンについて解析を行い,間接経路ニューロンが早期に失われやすいことが明らかにしました.この変化は舞踏運動の出現に関与している可能性があります.
また患者脳を細胞ごとに解析し,HDを引き起こすHTT遺伝子のCAGリピートが,病気の進行に伴い体細胞内で増加することを発見しました!とくに線条体のニューロンでは,初期にはリピートが安定しているものの,加齢とともに徐々に伸長し,特定の閾値を超えると急速に毒性を発揮することを示しました.この現象は「somatic expansion(体細胞伸長)」と呼ばれ,白血球DNAでは見られない脳特有の変化であることも確認されました.
注目すべき発見は,CAGリピートの数が150を超えるとニューロンが急速に傷害されるという点です.CAGリピートは通常は40程度ですが,時間が経過し80を超えると急速に拡大し,150,500を超えると神経毒性を持ちます.つまり「変異したHTTタンパク質の慢性的な毒性」ではなく,「リピート数が閾値を超えることで突然細胞が死ぬ」という新たなメカニズムを提唱するものです.これがニューロンの遺伝子発現を変化させ,細胞死を引き起こすということです.図で説明すると,「Phase A」では,体細胞内でCAGリピートが緩やかに拡張する.この段階は数十年という長い時間をかけて進行し,臨床的な症状は現れません.「Phase B」では,CAGリピートが急速に拡張し,短期間(数年)で150リピートを超える段階に達します.この急速な拡張が,遺伝子発現に影響を及ぼす準備段階となります.「Phase C」では,CAGリピートが500回以上に達し,これが原因で500以上の遺伝子発現が異常を来す段階に入ります.この段階は数か月という短期間で進行し,細胞内の転写機構や神経機能に深刻な影響を及ぼします.最終段階である「Phase D」では,脱抑制危機(de-repression crisis)が発生し,神経細胞が不可逆的に死に至ります.このプロセスにより,HDの特徴的な神経変性が完成します.図の下部は,年齢と線条体ニューロンの生存率の関係を示しています.
また研究では体細胞伸長は,DNA修復酵素(特にMSH3)による減少である可能性が高く,修復過程でCAGリピートが誤って伸長されると考えています.よってMSH3や関連する酵素をターゲットにしてCAGリピートの伸長を抑制することで,症状の発現を遅らせたり進行を抑えたりする治療法が考えられます.すでに発症している患者においても,まだ毒性閾値を超えていないニューロンに作用して病気の進行を遅らせる可能性があります.今回の研究は,HDの発症・進行における新しいメカニズムを示すだけでなく,将来の治療法開発に向けた重要な基盤となる可能性があります.
最初に紹介した3つの問題点について完全解明ではないかもしれませんが,大きな進展が得られました.CAGリピート病は私の学位論文のテーマですが,このようなメカニズムがあるとは夢にも思いませんでした.おそらくHD以外のCAGリピートでも同じことが起きているのではないかと思います.SCA6やCAGリピート以外のリピート病(例:筋強直性ジストロフィーや脆弱X症候群など)はどうなのかも気になります.近いうちにどんどんデータが報告されるのではないかと思います.
Handsaker, Robert E. et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease. Cell, January 16,(doi.org/10.1016/j.cell.2024.11.038)
研究チームへのインタビューです.
https://youtu.be/hd8Uukrocps