








さいたま市で行われた標題の学会にて,筑波大学皮膚科藤本学先生による講演を拝聴した.皮膚所見の見方について大変勉強になったのでまとめたい.
【皮膚所見の見方】
① 複数の好発部位をもれなく診察する
ヘリオトロープ疹やゴットロン徴候が有名であるが,皮膚筋炎にてこれら2つの徴候がいずれも認めない症例が約6%存在している.すなわちこれら2つの徴候のみでは診断ができないことになる.
また他の疾患で,ヘリオトロープ疹,ゴットロン徴候に類似した皮膚所見も呈しうる.前者は甲状腺機能低下症で類似の所見が見られ,後者は尋常性乾癬でも似た所見を呈しうる.
→ 以上2つの理由で,複数の好発部位をもれなく診察し,系統的に診断を進める必要がある.
② とくに「顔と耳と手指」を診察する
顔ではヘリオトロープ疹以外の皮疹にも注意する.鼻根部や鼻翼周囲(脂漏性皮膚炎様),前額部を確認する.通常の皮膚炎では所見を認めにくい耳の周囲にも皮疹が生じる.耳介の出っ張ったところに物理的刺激のために皮疹が生じやすい(ケブネル徴候).ちなみにSLEでは,寒冷刺激により耳朶に所見を呈する.
手では,爪の所見も重要である.SLEでは爪囲紅斑,強皮症では爪上皮出血点が見られるが,皮膚筋炎ではいずれも認められる.手背の丘疹状皮疹であるゴットロン丘疹や,丘疹状にならない紅斑であるゴットロン徴候が認められる.しかし,ゴットロン徴候にも様々な所見が認められる.角化が主体であるものは抗ARS抗体,丘疹性変化で炎症が強いものは抗TIF1抗体,滲出性紅斑や紫斑,潰瘍化の傾向があるものは抗MDA5抗体が陽性であることが多い.
「逆ゴットロン徴候」,すなわち関節屈側に,鉄棒を行った時にできるまめ様の所見(鉄棒まめ様所見)を認めることがある.この場合,抗MDA5抗体陽性であることが多い.一方,mechanic’s hand(機械工の手徴候)は,抗ARS抗体陽性例において認められる.
体幹では前胸部のV徴候や,肩から上背部のショール徴候,背中の掻爬による綿状の紅斑(むち打ち様紅斑)を認める.むち打ち様紅斑は掻爬によるケブネル徴候であり,診断的価値が高い.
【合併症による分類】
皮膚筋炎は「間接性肺炎と悪性腫瘍という2つの合併症」によって区別することができる.両者を合併することは稀である.
【抗体による分類】
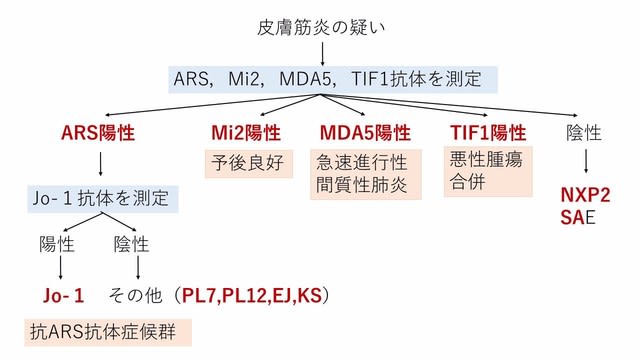
皮膚筋炎では70%以上の症例で陽性になる.1症例において1抗体のみ陽性となることから,分類に有用で,かつ抗体の種類と臨床像は強く相関する.頻度は成人と小児で異なり,成人の場合,抗ARS抗体>抗MDA5抗体>抗TIF1抗体>抗Mi2抗体>抗NXP2抗体>抗SAE抗体の順に多い.
① 抗ARS抗体(間接性肺炎は必発)
有名な抗Jo-1抗体を含む4つの抗体の総称であり,アミノアシルtRNA合成酵素(ARS)に対する抗体である(最初にJo-1抗体を測定するのではなく,まず抗ARS抗体を測定する).特徴として間接性肺炎はほぼ必発で,慢性の経過をとり,再燃を繰り返す.筋炎も再燃を繰り返す.皮膚はカサカサで,その他,レイノー現象や機械工の手徴候を認める.炎症症状を呈し,発熱,関節炎,CRP上昇を認める.CK上昇の程度はさまざま.
② 抗MDA5抗体(重症間質性肺炎タイプ)
ウィルスのRNA受容体でCADM(clinically amyopathic dermatomyositis)に多くみられる抗体.予後不良の急性進行性間質性肺炎を合併する.半数が重症で死亡例が多い.皮膚所見として逆ゴットロン徴候(鉄棒まめ様皮疹)を認める.これを認めた場合には,抗体の検査結果が出ていなくても治療を開始する,もしくは少なくとも準備を開始する.最初からステロイドに免疫抑制剤(タクロリムス,シクロフォスファミド)を併用する.IVIgは有効な印象がある.活動性のバイオマーカーとして,フェリチンや抗MDA5抗体価が有用である.CK値は2,000以下であることがほとんどである.
③抗TIF1抗体(悪性腫瘍タイプ)
悪性腫瘍を合併する.40歳以上の症例で70%に認める.皮疹が重症で,嚥下障害を合併することが多い.CK値はあまり高くなく,1000以下が多い.しかしCK値が高いほど,悪性腫瘍を合併することが多い.ステロイド抵抗性の嚥下障害に対してIVIgが有効である.
④抗Mi2抗体
筋症状の強い古典的皮膚筋炎の臨床像をとる.間質性肺炎はまれ.生命予後は悪くない.CK値は高めである.
⑤その他
抗NXP2抗体は,悪性腫瘍を合併する皮膚筋炎である.皮膚所見では石灰沈着が強い.
抗SAE抗体はTIF1抗体陽性例に症状が類似する.抗TIF1抗体陰性であればSAE抗体を疑う.
最後に皮膚筋炎を疑ったときの自己抗体の測定の手順について図に示す.
【皮膚所見の見方】
① 複数の好発部位をもれなく診察する
ヘリオトロープ疹やゴットロン徴候が有名であるが,皮膚筋炎にてこれら2つの徴候がいずれも認めない症例が約6%存在している.すなわちこれら2つの徴候のみでは診断ができないことになる.
また他の疾患で,ヘリオトロープ疹,ゴットロン徴候に類似した皮膚所見も呈しうる.前者は甲状腺機能低下症で類似の所見が見られ,後者は尋常性乾癬でも似た所見を呈しうる.
→ 以上2つの理由で,複数の好発部位をもれなく診察し,系統的に診断を進める必要がある.
② とくに「顔と耳と手指」を診察する
顔ではヘリオトロープ疹以外の皮疹にも注意する.鼻根部や鼻翼周囲(脂漏性皮膚炎様),前額部を確認する.通常の皮膚炎では所見を認めにくい耳の周囲にも皮疹が生じる.耳介の出っ張ったところに物理的刺激のために皮疹が生じやすい(ケブネル徴候).ちなみにSLEでは,寒冷刺激により耳朶に所見を呈する.
手では,爪の所見も重要である.SLEでは爪囲紅斑,強皮症では爪上皮出血点が見られるが,皮膚筋炎ではいずれも認められる.手背の丘疹状皮疹であるゴットロン丘疹や,丘疹状にならない紅斑であるゴットロン徴候が認められる.しかし,ゴットロン徴候にも様々な所見が認められる.角化が主体であるものは抗ARS抗体,丘疹性変化で炎症が強いものは抗TIF1抗体,滲出性紅斑や紫斑,潰瘍化の傾向があるものは抗MDA5抗体が陽性であることが多い.
「逆ゴットロン徴候」,すなわち関節屈側に,鉄棒を行った時にできるまめ様の所見(鉄棒まめ様所見)を認めることがある.この場合,抗MDA5抗体陽性であることが多い.一方,mechanic’s hand(機械工の手徴候)は,抗ARS抗体陽性例において認められる.
体幹では前胸部のV徴候や,肩から上背部のショール徴候,背中の掻爬による綿状の紅斑(むち打ち様紅斑)を認める.むち打ち様紅斑は掻爬によるケブネル徴候であり,診断的価値が高い.
【合併症による分類】
皮膚筋炎は「間接性肺炎と悪性腫瘍という2つの合併症」によって区別することができる.両者を合併することは稀である.
【抗体による分類】
皮膚筋炎では70%以上の症例で陽性になる.1症例において1抗体のみ陽性となることから,分類に有用で,かつ抗体の種類と臨床像は強く相関する.頻度は成人と小児で異なり,成人の場合,抗ARS抗体>抗MDA5抗体>抗TIF1抗体>抗Mi2抗体>抗NXP2抗体>抗SAE抗体の順に多い.
① 抗ARS抗体(間接性肺炎は必発)
有名な抗Jo-1抗体を含む4つの抗体の総称であり,アミノアシルtRNA合成酵素(ARS)に対する抗体である(最初にJo-1抗体を測定するのではなく,まず抗ARS抗体を測定する).特徴として間接性肺炎はほぼ必発で,慢性の経過をとり,再燃を繰り返す.筋炎も再燃を繰り返す.皮膚はカサカサで,その他,レイノー現象や機械工の手徴候を認める.炎症症状を呈し,発熱,関節炎,CRP上昇を認める.CK上昇の程度はさまざま.
② 抗MDA5抗体(重症間質性肺炎タイプ)
ウィルスのRNA受容体でCADM(clinically amyopathic dermatomyositis)に多くみられる抗体.予後不良の急性進行性間質性肺炎を合併する.半数が重症で死亡例が多い.皮膚所見として逆ゴットロン徴候(鉄棒まめ様皮疹)を認める.これを認めた場合には,抗体の検査結果が出ていなくても治療を開始する,もしくは少なくとも準備を開始する.最初からステロイドに免疫抑制剤(タクロリムス,シクロフォスファミド)を併用する.IVIgは有効な印象がある.活動性のバイオマーカーとして,フェリチンや抗MDA5抗体価が有用である.CK値は2,000以下であることがほとんどである.
③抗TIF1抗体(悪性腫瘍タイプ)
悪性腫瘍を合併する.40歳以上の症例で70%に認める.皮疹が重症で,嚥下障害を合併することが多い.CK値はあまり高くなく,1000以下が多い.しかしCK値が高いほど,悪性腫瘍を合併することが多い.ステロイド抵抗性の嚥下障害に対してIVIgが有効である.
④抗Mi2抗体
筋症状の強い古典的皮膚筋炎の臨床像をとる.間質性肺炎はまれ.生命予後は悪くない.CK値は高めである.
⑤その他
抗NXP2抗体は,悪性腫瘍を合併する皮膚筋炎である.皮膚所見では石灰沈着が強い.
抗SAE抗体はTIF1抗体陽性例に症状が類似する.抗TIF1抗体陰性であればSAE抗体を疑う.
最後に皮膚筋炎を疑ったときの自己抗体の測定の手順について図に示す.