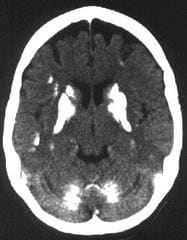
神経嚢虫症(neurocysticercosis)は有鉤条虫(Taenia solium)の幼虫である有鉤嚢尾虫(Cysticerus cellulosae)がCNSに達して引き起こされる疾患で,CNSの寄生虫感染症としては最も頻度が高い.感染ルートは主に豚肉(生,もしくは不完全に調理されたもの),虫卵に汚染された水や野菜である.MRI 上は多数の嚢胞がクモ膜下腔,実質内あるいは脳室内に認められ,内部に虫体を認めることも多い.嚢胞周囲に炎症・グリオーシスを伴っている場合には Gdにより輪状の増強効果が認められる.後期には石灰化を呈し,この時期にはMRIよりCTが有効になる.
今回,アメリカにおける神経嚢虫症の現状に関して,過去20年以上にわたるsummaryが報告された.具体的には1980年から2004年における神経嚢虫症の報告をPubMedより検索した.結果として1494例の症例がcase seriesにおいて報告されていた.うち76例がアメリカ国内での感染と考えられた.初発症状は多い順にてんかん(66%),水頭症(16%),頭痛(15%)であった.病変部位については脳実質が91%と大半を占めるが,そのほか脳室内嚢疱(6%),クモ膜下嚢疱(2%),脊髄(0.2%)も認められた.危険因子は,①流行地への旅行,②Hispanic系人種,③Taenia solium保因者との接触,であった.また文献報告数は増加の傾向にあり,アメリカでの本症の有病率増加が示唆された.
日本では稀な疾患ではあるが,中南米や東南アジア旅行者における感染例の報告はあり,本疾患について認識する必要がある.ちなみに治療には albendazole (ABZ) が有用であるが、病変が消失せず痙攣が続く場合には手術も行われる.
Neurology 63; 1559-1564, 2004
追伸;現地を離れるため,1月10日まで更新をお休みします.どうぞ良いお年を.
今回,アメリカにおける神経嚢虫症の現状に関して,過去20年以上にわたるsummaryが報告された.具体的には1980年から2004年における神経嚢虫症の報告をPubMedより検索した.結果として1494例の症例がcase seriesにおいて報告されていた.うち76例がアメリカ国内での感染と考えられた.初発症状は多い順にてんかん(66%),水頭症(16%),頭痛(15%)であった.病変部位については脳実質が91%と大半を占めるが,そのほか脳室内嚢疱(6%),クモ膜下嚢疱(2%),脊髄(0.2%)も認められた.危険因子は,①流行地への旅行,②Hispanic系人種,③Taenia solium保因者との接触,であった.また文献報告数は増加の傾向にあり,アメリカでの本症の有病率増加が示唆された.
日本では稀な疾患ではあるが,中南米や東南アジア旅行者における感染例の報告はあり,本疾患について認識する必要がある.ちなみに治療には albendazole (ABZ) が有用であるが、病変が消失せず痙攣が続く場合には手術も行われる.
Neurology 63; 1559-1564, 2004
追伸;現地を離れるため,1月10日まで更新をお休みします.どうぞ良いお年を.