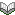最新号のNew Engl J Med誌に教育的な優れた総説が出ています.主旨は「PRES(posterior reversible encephalopathy syndrome)という名称はもはや疾患の特徴をすべて捉えているとは言えない」というものです.
まず画像所見に関して,症例集積研究では,疾患の特徴として知られる頭頂・後頭病変は65~99%ですが,その他の部位もかなり多く,前頭(54~88%),側頭(68%),視床(30%),小脳(34~53%),脳幹(18~27%),基底核(12~34%)なのだそうです.図のように両側大脳半球分水嶺(23%)(左),上前頭回(27%;中央),小脳半球(右)にも認めることもあります.よって診断は画像所見に頼るのではなく,病歴,つまり急激な高血圧や,PRESをきたす薬剤の使用歴(下記)が重要ということになります.Hincheyらが原著(1996)と比較すると,画像所見のみでなく,症候や誘発因子,非可逆的の予後までかなり逸脱した症例が存在します.著者は診断基準を作成するための学際的な協力体制の構築が必要と述べています.

【PRESをきたす薬剤一覧】
①化学療法薬:ベバシズマブ,チロシンキナーゼ阻害薬,ボルテゾミブ,シタラビン,ゲムシタビン, L-アスパラギナーゼ,メトトレキサート,ビンクリスチン,シスプラチン
②免疫調整剤:タクロリムス,シロリムス,シクロスポリンA,リツキシマブ,IL・TNF拮抗薬,インターフェロンα
③その他:エリスロポエチン,G-CSF,ボリコナゾール,偽エフェドリン,HIV感染症に対する抗レトロウイルス療法
④中毒:アルコール中毒,薬物の過剰摂取(リチウム,デキストロアンフェタミン,アセトアミノフェン,エフェドリン,フェニルプロパノールアミン,ジギトキシン,ビスマス),化学物質(例:有機リン酸エステル).違法薬物(コカイン,アンフェタミン,メフェドロン,クラトム,リゼルギン酸アミド),天然毒素(蛇・サソリ毒・スズメバチ毒,キノコ毒など)
Geocadin RG. Posterior Reversible Encephalopathy Syndrome. N Engl J Med. 2023 Jun 8;388(23):2171-2178.
少し前のNEJM誌のImage Challengeのクイズに出題されたこの画像もPRESでした.こちらの画像のほうがより分水嶺っぽく見えます.
「IgA腎症の35歳の男性が,錯乱,かすみ目,痙攣を呈した.受診の2週間前からシクロスポリンを投与していた.血圧160/80mmHg,眠気,視力低下を認めた」
N Engl J Med 2023; 388:e75

まず画像所見に関して,症例集積研究では,疾患の特徴として知られる頭頂・後頭病変は65~99%ですが,その他の部位もかなり多く,前頭(54~88%),側頭(68%),視床(30%),小脳(34~53%),脳幹(18~27%),基底核(12~34%)なのだそうです.図のように両側大脳半球分水嶺(23%)(左),上前頭回(27%;中央),小脳半球(右)にも認めることもあります.よって診断は画像所見に頼るのではなく,病歴,つまり急激な高血圧や,PRESをきたす薬剤の使用歴(下記)が重要ということになります.Hincheyらが原著(1996)と比較すると,画像所見のみでなく,症候や誘発因子,非可逆的の予後までかなり逸脱した症例が存在します.著者は診断基準を作成するための学際的な協力体制の構築が必要と述べています.
【PRESをきたす薬剤一覧】
①化学療法薬:ベバシズマブ,チロシンキナーゼ阻害薬,ボルテゾミブ,シタラビン,ゲムシタビン, L-アスパラギナーゼ,メトトレキサート,ビンクリスチン,シスプラチン
②免疫調整剤:タクロリムス,シロリムス,シクロスポリンA,リツキシマブ,IL・TNF拮抗薬,インターフェロンα
③その他:エリスロポエチン,G-CSF,ボリコナゾール,偽エフェドリン,HIV感染症に対する抗レトロウイルス療法
④中毒:アルコール中毒,薬物の過剰摂取(リチウム,デキストロアンフェタミン,アセトアミノフェン,エフェドリン,フェニルプロパノールアミン,ジギトキシン,ビスマス),化学物質(例:有機リン酸エステル).違法薬物(コカイン,アンフェタミン,メフェドロン,クラトム,リゼルギン酸アミド),天然毒素(蛇・サソリ毒・スズメバチ毒,キノコ毒など)
Geocadin RG. Posterior Reversible Encephalopathy Syndrome. N Engl J Med. 2023 Jun 8;388(23):2171-2178.
少し前のNEJM誌のImage Challengeのクイズに出題されたこの画像もPRESでした.こちらの画像のほうがより分水嶺っぽく見えます.
「IgA腎症の35歳の男性が,錯乱,かすみ目,痙攣を呈した.受診の2週間前からシクロスポリンを投与していた.血圧160/80mmHg,眠気,視力低下を認めた」
N Engl J Med 2023; 388:e75