がんの予防や治療における漢方治療の存在意義を考察しています。がん治療に役立つ情報も紹介しています。
「漢方がん治療」を考える
カレンダー
| 2024年9月 | ||||||||
| 日 | 月 | 火 | 水 | 木 | 金 | 土 | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| 8 | 9 | 10 | 11 | 12 | 13 | 14 | ||
| 15 | 16 | 17 | 18 | 19 | 20 | 21 | ||
| 22 | 23 | 24 | 25 | 26 | 27 | 28 | ||
| 29 | 30 | |||||||
|
||||||||
goo ブログ
過去の記事
カテゴリ
最新の投稿
最新のコメント
最新のトラックバック
ブックマーク
プロフィール
検索
gooおすすめリンク




| URLをメールで送信する | |
| (for PC & MOBILE) | |
716)プテロスチルベン(Pterostilbene)の抗がん作用(その1):JAK/STAT3シグナル伝達系とPI3K/AKTシグナル伝達系の阻害
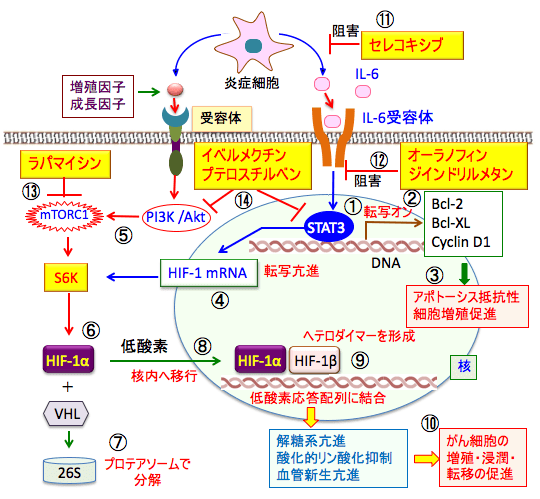
図:活性化した炎症細胞から産生される炎症性サイトカインのIL-6はがん細胞のIL-6受容体を介してSTAT3を活性化し(①)、Bcl-2やBcl-XLやCyclin D1の転写を亢進し(②)、アポトーシス抵抗性や細胞増殖を促進する(③)。さらに、低酸素誘導因子-1(HIF-1)の転写を亢進する(④)。増殖因子や成長因子によるシグナルはPI3K/Aktシグナル伝達系を介してmTORC1(哺乳類ラパマイシン標的タンパク質複合体1)を活性化する(⑤)。mTORC1はリボソームの生合成を促進するS6Kをリン酸化して活性化する作用によって蛋白質合成を促進し、HIF-1タンパク質の産生を増やす(⑥)。酸素濃度が高い状態では、HIF-1αは酸素濃度感受性タンパク質のプロリル・ヒドロキシラーゼによって水酸化され、VHL(von Hippel-Lindau)タンパク質が結合して26Sプロテアソームで分解される(⑦)。低酸素状態ではプロリル・ヒドロキシラーゼの活性が低下してHIF-1αの分解が阻止されるので、蓄積したHIF-1αは核内に移行し(⑧)、HIF-1βとヘテロダイマー(ヘテロ二量体)を形成して遺伝子の低酸素応答配列に結合し、低酸素状態の適応に必要な様々な遺伝子の発現を誘導する(⑨)。その結果、解糖系が亢進し、酸化的リン酸化が抑制され、血管新生を促進されて、がん細胞の増殖・浸潤・転移が促進される(⑩)。シクロオキシゲナーゼ-2(COX-2)阻害剤のセレコキシブ(celecoxib)は炎症細胞の活性を抑制し(⑪)、オーラノフィンとジインドリルメタンはSTAT3の活性化を抑制する(⑫)。ラパマイシンはmTORC1を直接的に阻害する(⑬)。イベルメクチンとプテロスチルベンはSTAT3経路とPI3K/Akt経路の両方を阻害する(⑭)。これらの医薬品を組み合せると、がん細胞の増殖を抑制する効果が期待できる。
716)プテロスチルベン(Pterostilbene)の抗がん作用(その1):JAK/STAT3シグナル伝達系とPI3K/AKTシグナル伝達系の阻害
【プテロスチルベンは植物が合成する抗菌物質(ファイトアレキシン)の一つ】
植物は、外敵(病原菌など)や過酷な外的環境(紫外線や熱や重金属など)に打ち勝つために、様々な生体防御物質を合成しています。植物体に病原菌や寄生菌が侵入したときに植物細胞が合成する抗菌性物質をファイトアレキシン(phytoalexin)と言います。
刺激を受けて合成を開始しますが、すでに持っている物質を活性化することもあります。多くの化合物が知られていますが、ブドウやブルーベリーなどに含まれるレスベラトロール(Resveratrol)もファイトアレキシンの一つです。
プテロスチルベン(Pterostilbene)はレスベラトロールの類縁体で、レスベラトロールの2つの水酸基(OH)がメトキシ基(CH3O-)に置換した構造です。
プテロスチルベンはレスベラトロールと同様にブドウやブルーベリーなどに含まれます。
プテロスチルベンはレスベラトロールに比べて生体利用率が極めて高く、生体に対する薬効もプテロスチルベンの方が優れていることは前回(715話)解説しています。

図:プテロスチルベンはレスベラトロールの2個の水酸基(OH)がメトキシ基(CH3O-)に置換している。2つのメトキシ基が存在することで、プテロスチルベンはより親油性になり、消化管からの吸収がよく、グルクロン酸抱合や硫酸抱合による不活性化を受けにくいので、生物学的利用能がレスベラトロールより高い。
プテロスチルベンが様々ながん細胞において、PI3K / AKT経路、MAPK経路、JAK/STAT経路、Notch経路、Wnt経路など様々なシグナル伝達経路や、転移関連タンパク質1(MTA1)、ヒトテロメラーゼ逆転写酵素(hTERT)など、がん細胞の増殖や生存に関与する多くのシグナル伝達系やタンパク質に作用して抗腫瘍活性を発揮することが報告されています。
JAK/STAT3シグナル伝達系とPI3K/AKTシグナル伝達系の阻害作用について解説します。
【STAT3はがん細胞の増殖を促進する】
STAT3は、STAT (Signal Tranducer and Activator of Transcription:シグナル伝達兼転写活性化因子) ファミリーに属する蛋白質で、その名の通り、「シグナル伝達」と「遺伝子転写活性化」の両方において働きます。
STAT3は非活性化状態においては細胞質に存在しますが、ヤーヌスキナーゼ(Janus Kinase; JAK)が活性化されることによってリン酸化を受け、核内へ移行して目的遺伝子を活性化する転写因子として機能します。
JAK(ヤーヌスキナーゼ)はサイトカイン受容体のサブユニットとして存在し、チロシンをリン酸化する酵素(チロシンキナーゼ)の一種です。
IL-6ファミリーのサイトカインあるいは上皮成長因子(EGF)等の成長因子がそれらの受容体に結合することによりヤーヌスキナーゼ(JAK)が活性化されると、活性化されたJAKがSTAT3のチロシン705をリン酸化します。
チロシン705がリン酸化されたSTAT3二分子のSH2ドメインがそれぞれ他方の分子のリン酸化チロシンと相互作用することにより二量体を形成して核内に移行し、核内に移行したSTAT3二量体は標的となるDNAに結合する事で転写を活性化します。これをJAK-STAT経路と言います。
STAT3のリン酸化はJAKを介する以外に、増殖因子や成長因子の受容体が直接リン酸化する場合や、Srcなどの非受容体性チロシン・キナーゼによっても起こります。つまり、様々な細胞刺激に応答してSTAT3がリン酸化されて、増殖や生存を促進する作用を発揮します(下図)。

図:JAK(Janus Kinase;ヤーヌスキナーゼ)はサイトカイン受容体のサブユニットとして存在し、チロシンをリン酸化するチロシンキナーゼ活性を持つ(①)。IL-6や上皮成長因子(EGF)などの受容体が刺激されるとJAKが活性化されてSTAT3がリン酸化される(②)。STAT3のリン酸化は受容体性チロシンキナーゼや非受容体性チロシンキナーゼ(Srcなど)でも起こる(③)。STAT3は不活性な状態では細胞質に存在し、JAK(ヤーヌスキナーゼ)などでチロシン705がリン酸化されると、STAT3二分子のSH2ドメインが、それぞれ他方の分子のリン酸化チロシンと相互作用することにより二量体を形成して核内に移行する(④)。核内に移行したSTAT3二量体は、標的となるDNAに結合する事で転写を活性化する(⑤)。STAT3は細胞をアポトーシス抵抗性にするBcl-2やBcl-XL、細胞周期を促進するサイクリンD1(Cyclin D1)などの遺伝子発現を誘導することによってがん細胞の増殖や転移を促進する(⑥)。
【多くのがん細胞はPI3K / Akt経路とERK-MAPK経路が活性化している】
細胞の増殖は、増殖因子受容体が細胞外ドメインで増殖シグナルを受け取ることから始まります。
細胞内で機能している多数のシグナル伝達経路の中で、がん細胞の増殖と生存で最も重要なのが、PI3K-Akt経路(生存シグナル経路)とERK-MAPK経路(増殖シグナル経路)です。
細胞膜の増殖因子受容体にリガンド(増殖因子)が結合し2量体化すると、PI3Kのリン酸化活性からAktのリン酸化を通して、アポトーシス(細胞死)の誘導を阻害します。(PI3K-Akt経路)
増殖因子による刺激は、低分子量G蛋白質Rasを経由して、Raf→MEK→ERKとリン酸化反応するMAPK経路(MAPKカスケード)によりシグナルが伝達されます。活性化したERKは最終的に核へ移行し、転写因子が活性化され、細胞増殖関連の遺伝子が発現します。(ERK-MAPK 経路)
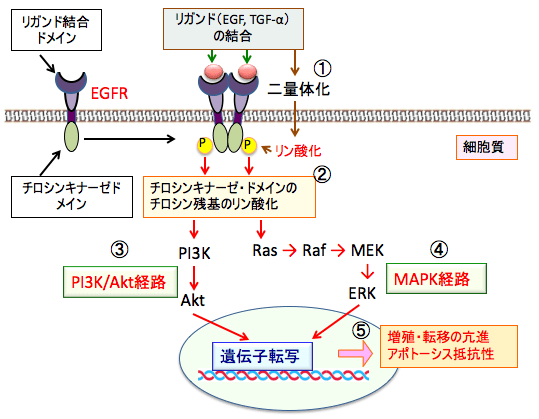
図:チロシンリン酸化型受容体にリガンド(増殖因子や成長因子)が結合し2量体化すると(①)、受容体がリン酸化されて活性化する(②)。受容体が活性化されるとPI3Kのリン酸化活性からAktがリン酸化されて活性化する。これをPI3K/Akt経路という(③)。一方、受容体の活性化は、低分子量G蛋白質Rasを経由して、Raf→MEK→ERKとリン酸化反応するMAPK経路(④)によりシグナルが伝達される。PI3K/Akt経路とMAPK経路の活性化は、最終的に核の転写因子の活性化を介して、がん細胞の増殖や転移を亢進し、アポトーシスに抵抗性(死ににくくなる)の性質を持つようになる(⑤)。
Akt (プロテインキナーゼBまたはPKBとしても知られている) はセリン/スレオニンキナーゼです。多くのシグナル伝達経路のネットワークの中心的存在で、下流の幅広いターゲット分子や相互作用分子を介してさまざまな細胞内反応を引き起こします。
Aktは、CDK阻害因子のp21及びp27に対する直接作用や、サイクリンD1 (Cyclin D1) 及びp53のレベルに対する間接的な作用によって細胞周期と細胞分裂を調節するとともに、mTORとp70 S6キナーゼ経路に対する効果を通じて細胞増殖を調節します。
Aktは、BadやForkheadファミリーの転写因子のようなアポトーシス促進性のシグナルを直接阻害することによって、細胞の生存を媒介する主要なメディエーターです。
したがって、Aktの活性化を阻害することはがん細胞の増殖を抑制し、細胞死(アポトーシス)を誘導します。
PI3K-Akt経路は、乳がん、卵巣がん、大腸がん、前立腺がん、神経膠芽腫など多くの腫瘍において恒常的機能亢進が認められています。PI3K-Akt経路の活性抑制は多くのがん細胞の増殖や転移を抑制します。

図:PI3キナーゼ(PI3K)-AKT経路は,細胞外からのシグナルを細胞内に伝える主要な経路の一つで、増殖因子やインテグリンを介した細胞接着など、様々な刺激により活性化され、細胞の生存促進,細胞増殖・大きさの制御、細胞運動、代謝の制御など多くの細胞機能に関与している。ホスファチジルイノシトール3-キナーゼ(PI3K)は,細胞膜の構成成分であるイノシトールリン脂質をリン酸化する酵素で,産生したPI3,4,5-三リン酸(PIP3)がAktをリン酸化する。Aktは多くのがん組織で活性しておりAKTシグナル伝達系は、細胞増殖や生存、細胞サイズや栄養素利用への応答性、グルコース代謝、組織浸潤および血管新生など多くの細胞プロセスを制御している。がん細胞の生存と増殖はAKTシグナル伝達系の活性に依存度が高いので、AKTシグナル伝達系の阻害はがん細胞の増殖抑制やアポトーシス誘導を引き起こす。
ホスファチジルイノシトール3-キナーゼ(PI3K)は,細胞膜の構成成分であるイノシトールリン脂質をリン酸化する酵素です。ホスファチジルイノシトール4,5-二リン酸(PIP2)の3位のOHをリン酸化してホスファチジルイノシトール3,4,5-三リン酸(PIP3)を生成する酵素です。PIP3がAktをリン酸化して活性化します。
PTENは脱リン酸化する酵素でPIP3をPIP2に変化することによってAktの活性化を阻止します。

図:ホスファチジルイノシトール3-キナーゼ(PI3K)は,細胞膜の構成成分であるイノシトールリン脂質をリン酸化する酵素で,産生したPI3,4,5-三リン酸(PIP3)がAktをリン酸化する。Aktは多くのがん組織で活性しておりAKTシグナル伝達系は、細胞増殖や生存、細胞サイズや栄養素利用への応答性、グルコース代謝、組織浸潤および血管新生など多くの細胞プロセスを制御している。PTENはPIP3を脱リン酸化してPIP2に変換し、Akt活性化を阻止する。
PI3Kはp85αとp110の2つのサブユニットから構成されています。
PI3K-Aktシグナル伝達系は細胞の生存と増殖の制御の中心です。活性化したAktがシグナル伝達の下流に位置する様々なタンパク質をリン酸化して生存や増殖を制御しています。

図:セリン/スレオニンキナーゼのAktは、多くのシグナル伝達経路のネットワークの中心的存在で、下流の幅広いターゲット分子や相互作用分子を介してさまざまな細胞内反応を引き起こす。
【HIF-1は低酸素になると活性化される】
細胞内での情報シグナル伝達系の方法は主にリン酸化です。ATPを用いてリン酸化のスイッチのONとOFFを繰り返して情報を伝達します。このような情報伝達のネットワークはクロストーク(他の伝達経路と影響しあうこと)します。
PI3K/AKT経路とJAK/STAT3経路はクロストークします。その一つの例が低酸素誘導因子-1(HIF-1)の制御です。
生物は外界の酸素濃度を認識する巧みな仕組みを保持しています。
酸素濃度が低下すると、生物は低酸素シグナルを活性化し低酸素状態に適応します。
この低酸素応答の中心的分子が低酸素誘導因子-1(Hypoxia inducible factor-1: HIF-1) およびプロリル・ヒドロキシラーゼ(prolyl hydroxylase)と呼ばれるタンパク質です。
HIF-1は、細胞が酸素不足に陥った際に誘導されてくる転写因子です。αとβの2つのサブユニットからなるヘテロ二量体であり、βサブユニットは定常的に発現して細胞核にいますが、 HIF-1αは細胞質で酸素濃度依存的な分解を受けます。(HIFのαサブユニットにはHIF-1α, -2α and -3α、βサブユニットにはHIF-1β, -2β and -3βのそれぞれ3種類が知られていますが、低酸素誘導因子として中心になっているのはHIF-1αとHIF-1βであるため、HIF-1をHIFの同義語として使用)
すなわち、HIF-1αは、正常酸素濃度下では、HIF-1αタンパク質中の2カ所のプロリン残基がプロリル・ヒドロキシラーゼにより水酸化されることによりVHL(von Hippel-Lindau)タンパク質が結合します。
VHLが結合するとHIF-1αのユビキチン化が促進されて26Sプロテアソームで分解されます。したがって、酸素が十分にある状況ではHIF-1は不活性の状態に維持されます。
VHLはフォンヒッペル・リンドウ(von Hippel-Lindau)病の原因遺伝子として発見されています。
フォンヒッペル・リンドウ病は常染色体優性遺伝疾患で、脳・脊髄・網膜の血管芽腫や腎臓がんや褐色細胞腫などの腫瘍が多発します。
VHLたんぱく質が欠損するとHIF-1αが分解されないので、HIF-1転写活性が亢進した状態になり、VEGF(血管内皮細胞増殖因子)のタンパク量が増え、血管の豊富な腫瘍が発生します。
プロリル・ヒドロキシラーゼ(prolyl hydroxylase)は酸素濃度感受性のタンパク質で、酸素濃度が低下するとプロリル・ヒドロキシラーゼの酵素活性が著しく低下します。すると、HIF-1αのプロリン残基の水酸化が起きないので、HIF-1αは分解を受けずに安定化します。
安定化したHIF-1αは核内に移行し、HIF-1βと二量体を形成して低酸素応答配列(Hypoxia Responsive Element)に結合して、低酸素応答に必要な様々な遺伝子の発現を活性化します。
すなわち、HIF-1は各種解糖系酵素、グルコース輸送蛋白、血管内皮細胞増殖因子(VEGF)、造血因子エリスロポイエチンなど、 多くの遺伝子の発現を転写レベルで制御し、細胞から組織・個体にいたる全てのレベルの低酸素適応反応を制御しています(下図)。

図:酸素濃度が高い状態では、HIF-1αは酸素濃度感受性タンパク質のプロリル・ヒドロキシラーゼによって水酸化され(①)、VHL(von Hippel-Lindau)タンパク質が結合して26Sプロテアソームで分解される(②)。低酸素状態ではプロリル・ヒドロキシラーゼの活性が低下してHIF-1αの分解が阻止されるので、蓄積したHIF-1αは核内に移行し(③)、HIF-1βとヘテロダイマー(ヘテロ二量体)を形成して遺伝子の低酸素応答配列に結合し(④)、コアクチベーター(CBP/p300)やRNAポリメラーゼがリクルートされて遺伝子転写を亢進し(⑤)、低酸素状態の適応に必要な様々な遺伝子の発現を誘導する(⑥)。
【がん細胞では低酸素でなくてもHIF-1が恒常的に活性化している】
がん細胞の代謝の特徴は、酸素が十分に利用できる状況でも、酸素を使わない解糖系が亢進し、ミトコンドリアでの酸素を使ったエネルギー産生(酸化的リン酸化)が抑制されていることです。つまり、酸素があっても、あたかも低酸素のような代謝を行っているわけです。
このような代謝の特徴の根本的なメカニズムは、がん細胞では酸素濃度とは関係なく、恒常的にHIF-1が活性化しているためです。つまり、がん細胞では恒常的に低酸素シグナルがオンになっているということです。その理由は、がん細胞で活性化されているmTORC1やSTAT3がHIF-1の産生を促進するからです。
がん細胞の増殖シグナル伝達系であるPI-3キナーゼ/Akt/mTORC1シグナル伝達系においてmTORC1(哺乳類ラパマイシン標的タンパク質複合体1)は、リボソームの生合成を促進するS6Kをリン酸化して活性化する作用によって蛋白質合成(mRNAからタンパク質の翻訳)を促進し、HIF-1タンパク質の産生を増やします。
また、増殖因子やサイトカインで活性化されるSTAT3という転写因子はHIF-1遺伝子の転写を亢進します。
つまり、がん細胞で活性が亢進しているPI-3キナーゼ/Akt/mTORC1シグナル伝達系とJAK/STAT3はHIF-1タンパク質の産生量を相乗的に高めることが報告されています(下図)。

図:増殖刺激や遺伝子変異などによってがん細胞で恒常的に活性が亢進しているSTAT3(シグナル伝達兼転写活性化因子)はHIF-1遺伝子の転写(mRNAの産生)を促進し、mTORC1はリボソームの生合成を促進するS6Kを活性化してHIF-1タンパク質の合成を促進する。
HIF-1αタンパク質の発現量が増えても、HIF-1αの分解に関与するプロリル・ヒドロキシラーゼ(prolyl hydroxylase)やVHL(von Hippel-Lindau)タンパク質が正常に働けばHIF-1の活性亢進を抑制できますが、がん細胞ではプロリル・ヒドロキシラーゼやVHLの発現低下や遺伝子変異によってHIF-1αの分解過程に異常を起こしていることが多いので、HIF-1αは恒常的に高いレベルに亢進しています。
STAT-3を阻害する物質として、オーラノフィン、ジインドリルメタン、セレコキシブがあります。(427話、428話参照)
mTORC1の阻害剤としてラパマイシンがあります。(383話参照)
つまり、HIF-1αの発現と活性を阻害する方法として、オーラノフィン、ジインドリルメタン、セレコキシブ、ラパマイシンの組合せは有効かもしれません。メラトニンもHIF-1の活性化を阻害するという報告があります。
【PAK1はSTAT3の転写活性を促進する】
転写因子のSTAT3はほとんどすべての固形がんで活性化されており、細胞のがん化に重要な働きを担っていることが明らかになっています。活性化されたSTAT3が核内に移行するメカニズムに、低分子量G蛋白質Rac1の関与が報告されています。
PAK1(p21活性化キナーゼ1)はRacおよびCdc42のような低分子量GTPaseによって活性化されます。
乳がん幹細胞形成にPAK1-STAT3経路が重要で、この経路をPAK1阻害剤のイベルメクチンが阻害できることが報告されています。以下のような報告があります。
The PAK1-Stat3 Signaling Pathway Activates IL-6 Gene Transcription and Human Breast Cancer Stem Cell Formation(PAK1-Stat3シグナル伝達経路はIL-6遺伝子の転写とヒト乳がん幹細胞形成を活性化する)Cancers (Basel). 2019 Oct; 11(10): 1527.
【要旨】
がん幹細胞は、自己複製、分化、化学療法抵抗性などのユニークな特性を有する。この研究では、p21活性化キナーゼ(PAK1)阻害剤(IPA-3)およびPAK1不活性化剤(イベルメクチン)の投与が細胞増殖を阻害し、さらにマウスを用いた実験モデルで、PAK1遺伝子の阻害が腫瘍成長を有意に阻害した。
PAK1活性を阻害するIPAK-3とイベルメクチンはがん幹細胞の形成を阻害した。
PAK1は物理的にヤーヌスキナーゼ2(Janus Kinase 2:JAK2)と相互作用し、ヤーヌスキナーゼ2阻害剤は乳がん細胞塊の形成を阻害し、核におけるPAK1タンパク質レベルを低下させる。
PAK1は、シグナル伝達兼転写活性化因子-3(Stat3)と相互作用し、PAK1とStat3は核内で共局在している。
電気泳動移動度シフトアッセイ、クロマチン免疫沈降、およびレポーターアッセイを用いた解析で、PAK1 / Stat3複合体がIL-6遺伝子プロモーターに結合し、IL-6遺伝子の転写を制御することを示す。
乳がん細胞塊でのPAK1およびヤーヌスキナーゼ2(JAK2)の阻害は、核のリン酸化Stat3および細胞外IL-6レベルを低下させる。
PAK1不活性化は、リン酸化STAT3および細胞外IL-6レベルを低下させることにより、がん幹細胞形成を阻害する
これらの実験結果は、JAK2 / PAK1経路の阻害がStat3シグナル伝達経路とがん幹細胞形成を阻害し、PAK1 / Stat3複合体がIL-6遺伝子発現を調節し、PAK1 / Stat3シグナル伝達ががん幹細胞形成を調節し、PAK1が乳がん治療の重要な標的である可能性があることを明らかにしている。
この論文の結果をまとめると以下のようになります。

図: STAT3(シグナル伝達兼転写活性化因子-3)はヤーヌスキナーゼ2(JAK2)によってリン酸化されると、二量体を形成して核内に移行する(①)。核内に移行したSTAT3二量体は、標的となるIL-6遺伝子のプロモーター領域に結合する事でIL-6の転写を活性化する(②)。JAK2によってリン酸化して活性化したPAK1はSTAT3二量体に結合して遺伝子転写活性を促進する(③)。産生されたIL-6は細胞外に移行し(④)、さらにIL-6受容体を刺激し、このJAK2/PAK1/Stat3/IL-6経路ががん幹細胞の形成と幹細胞特性の維持に働く(⑤)。イベルメクチンはPAK1の活性化を阻害し(⑥)、IL-6遺伝子の転写を阻害する(⑦)。(参考:Cancers (Basel). 2019 Oct; 11(10): 1527.)
PAK1は物理的にJAK2と相互作用し、PAK1-JAK2がSTAT3の活性化を介してIL-6遺伝子の発現を亢進し、乳がんの幹細胞形成を制御しているという結果です。
STAT3がIL-6遺伝子のプロモーター領域に結合して、IL-6の発現を亢進します。
乳がん幹細胞の細胞質と核にPAK1が高度に発現し、全体に分布しています。通常のがん細胞に比べて乳がん幹細胞にPAK1の発現が亢進していることが明らかになっています。
そこで、PAK1活性を阻害すると、がん細胞の増殖が阻害されます。動物実験でもPAK1の阻害は腫瘍の増大を抑制します。
PAK1とSTAT3はがん幹細胞に重要で、この2つを阻害すれば、乳がん幹細胞の活性を抑制できるという結果です。
PAK1阻害剤のイベルメクチンはがん幹細胞のマーカー(CD44とALDH)をもつがん細胞の数を減らすことを明らかにしています。イベルメクチンはPAK1阻害作用によってSTAT3活性を抑制し、がん幹細胞の活性を低下します。
つまり、乳がんの幹細胞の形成においてPAK1/STAT3経路の活性化が重要で、PAK1阻害作用のあるイベルメクチンは乳がんの幹細胞形成を阻害するということです。

図: STAT3(シグナル伝達兼転写活性化因子-3)は受容体性および非受容体性のチロシンキナーゼによってリン酸化されると、二量体を形成して核内に移行する(①)。核内に移行したSTAT3二量体は、標的となるIL-6遺伝子のプロモーター領域に結合する事でIL-6の転写を活性化する(②)。JAK2によってリン酸化して活性化したPAK1はSTAT3二量体に結合して遺伝子転写活性を促進する(③)。産生されたIL-6は細胞外に移行し(④)、さらにIL-6受容体を刺激し、このJAK2/PAK1/Stat3/IL-6経路(回路)ががん幹細胞の形成と幹細胞特性の維持に働く(⑤)。イベルメクチンはPAK1の活性化を阻害し(⑥)、オーラノフィンとジインドリルメタンはSTAT3活性化を阻害する(⑦)。その結果、がん幹細胞の特性維持を阻止できる。(参考:Cancers (Basel). 2019 Oct; 11(10): 1527.)
イベルメクチンはSTAT3経路の阻害に加えて、Akt/mTORC1経路やWnt/βカテニン経路も阻害します。(674話参照)

図:低分子量Gタンパク質のRASは細胞外のさまざまな刺激(チロシンキナーゼ受容体やサイトカイン受容体やカルシウムチャネルなど)を受けて細胞増殖や生存を促進するRAFキナーゼ(Raf-1)やPI-3キナーゼ(PI3K)など多数のシグナル伝達系を活性化する(①)。p21活性化キナーゼ(PAK-1)はRacおよびCdc42のような低分子量GTPaseによって活性化される(②)。PAK-1はRasシグナル伝達系の主要な経路であるRaf-1/MEK1/ERK経路(③)とPI3K/AKT経路(④)を活性化する。さらに、PAK-1はβ-カテニンをリン酸化してWNT/β-カテニン経路を活性化する(⑤)。このように、PAK-1は複数のシグナル伝達系の制御に関与し、がん細胞の増殖と転移を促進する(⑥)。寄生虫治療薬のイベルメクチン(Ivermectin)はPAK-1とWNT/β-カテニン経路を阻害する作用によって、抗腫瘍作用を発揮する(⑦)。
プテロスチルベンはJAK/STAT3シグナル伝達系やPI3K/AKTシグナル伝達系の阻害作用が報告されています。MAPK経路、Notch経路、Wnt経路など様々なシグナル伝達経路の阻害作用も報告されています。
がん細胞の増殖や生存を促進するシグナル伝達系はお互いにネットワークを形成しています。したがって、1つの薬で1つのターゲットを阻害しても、多くの場合、ネットワークの迂回路を使って、増殖と生存を維持します。
イベルメクチンやプテロスチルベンは複数のシグナル伝達系を阻害しますが、これらだけでは抗腫瘍効果は限定的です。さらに異なるメカニズムでの増殖と生存のシグナル伝達系の阻害を併用することも重要です。
JAK/STAT3シグナル伝達系やPI3K/AKTシグナル伝達系やMAPK経路などの阻害をターゲットにしたがん治療として、セレコキシブ(celecoxib)、オーラノフィン、ジインドリルメタン、ラパマイシン、イベルメクチン、プテロスチルベンなどの組み合わせは効果が期待できるかもしれません。(トップの図参照)
さらに、解糖系を阻害する2-デオキシグルコースや、メトホルミンやケトン食などで、がん細胞の代謝やエネルギー産生を阻害する方法を併用すると、さらにがん細胞の増殖を抑制できると思います。
メトホルミンがCOX-2阻害剤のセレコキシブの抗腫瘍効果を増強することが最近報告されています。以下のような論文があります。
Metformin Synergistically Enhanced the Antitumor Activity of Celecoxib in Human Non-Small Cell Lung Cancer Cells.(メトホルミンは、ヒト非小細胞肺がん細胞においてセレコキシブの抗腫瘍活性を相乗的に増強した)Front Pharmacol. 2020; 11: 1094.
【要旨】
セレコキシブは有効な抗腫瘍薬としての可能性を秘めているが、副作用を示す可能性がある。がん細胞はグルコース依存性の特性を有し、メトホルミンはミトコンドリアにおける酸化的リン酸化を阻害する。
本研究では、低用量のセレコキシブとメトホルミンを組み合わせて、非ステロイド性抗炎症薬(NSAID)の副作用を緩和し、潜在的な薬剤耐性を克服することを目的とした。
セレコキシブとメトホルミンを組み合わせると、がん細胞の移動と増殖が明らかに抑制され、アポトーシスが誘導されることが明らかになった。
最も重要なことに、in vivoの実験では、低用量のセレコキシブ(25 mg / kg /日)とメトホルミンの併用は、明らかな毒性なしに明らかな抗腫瘍効果発揮した。
作用メカニズムのさらなる研究により、2つの薬物の組み合わせが非小細胞性肺がん細胞で活性酸素種の産生を引き起こし、DNA二本鎖の切断と腫瘍抑制因子p53の発現の増加を引き起こすことが明らかになった。 p53の上昇は、細胞周期の停止と細胞増殖の阻害を引き起こした。
メトホルミンの存在は、カスパーゼ-9、-8、-3、および-7を活性化し、アポトーシス促進タンパク質BadおよびBaxの発現を亢進し、抗アポトーシスタンパク質Bcl-xlおよびBcl-の発現を抑制することにより、非小細胞性肺がん細胞におけるセレコキシブ誘発アポトーシスの感受性を高めた。
さらに、併用療法の優れた抗がん効果は、Raf-MEK-ERKカスケードとPI3K-AKTシグナル伝達の抑制によるものであり、薬剤耐性の克服に貢献していた。
さらに、セレコキシブ単独またはセレコキシブ+メトホルミンの組み合わせは、FAK、N-カドヘリン、およびマトリックスメタロプロテイナーゼ-9の活性を阻害することにより、非小細胞性肺がん細胞の遊走および浸潤を抑制した。
以上の結果から、私たちの研究は、低用量のセレコキシブとメトホルミンの組み合わせががん治療における合理的な戦略を提供する可能性を示している。
この論文では、セレコキシブとメトホルミンが、Raf-MEK-ERKカスケードとPI3K-AKTシグナル伝達系の阻害において相乗的に作用する可能性を報告しています。
つまり、セレコキシブ+メトホルミンに、プテロスチルベン、イベルメクチン、オーラノフィン、ジインドリルメタンなどを併用すると、さらに抗がん作用を強化できると思います。
これらはいずれも副作用は極めて軽度で、比較的安価です。
● プテロスチルベンのサプリメントについてはこちらへ:


画像をクリックするとサイトに移行します
| « 715)レスベラ... | 717)プテロス... » |