








総説 血管内皮細胞のTRPA1について
You have free access to this contentTRPA1 channels in the vasculature (pages 13–22)
Scott Earley
Article first published online: 3 AUG 2012 | DOI: 10.1111
Abstruct
This review is focused on the role of the ankyrin (A) transient receptor potential (TRP) channel TRPA1 in vascular regulation. TRPA1 is activated by environmental irritants, pungent compounds found in foods such as garlic, mustard and cinnamon, as well as metabolites produced during oxidative stress. The structure of the channel is distinguished by the ∼14–19 ankyrin repeat (AR) domains present in the intracellular amino terminus. TRPA1 has a large unitary conductance (98 pS) and slight selectivity for Ca2+ versus Na+ ions (PCa/PNa ≈ 7.9). TRPA1 is involved in numerous important physiological processes, including nociception, mechanotransduction, and thermal and oxygen sensing. TRPA1 agonists cause arterial dilation through two distinctive pathways. TRPA1 channels present in perivascular nerves mediate vasodilatation of peripheral arteries in response to chemical agonists through a mechanism requiring release of calcitonin gene-related peptide. In the cerebral circulation, TRPA1 channels are present in the endothelium, concentrated within myoendothelial junction sites. Activation of TRPA1 channels in this vascular bed causes endothelium-dependent smooth muscle cell hyperpolarization and vasodilatation that requires the activity of small and intermediate conductance Ca2+-activated K+ channels. Systemic administration of TRPA1 agonists causes transient depressor responses, followed by sustained increases in heart rate and blood pressure that may result from elevated sympathetic nervous activity. These findings indicate that TRPA1 activity influences vascular function, but the precise role and significance of the channel in the cardiovascular system remains to be determined.
Transient Receptor Potential superfamily について
The mammalian transient receptor potential (TRP) superfamily of cation channels comprises 28 members assigned to six subfamilies based on sequence homology. The ankyrin (A) subfamily is the smallest and is composed of only a single member, TRPA1 (originally designated as ANKTM1). Despite being one of the last TRP channels to be discovered (Story et al., 2003), TRPA1 has garnered a great deal of recent attention. The essential properties and structure of TRPA1 have been evolutionarily conserved for more than 500 million years (Kang et al., 2010). The channel likely evolved as a sensor of electrophilic toxicity (Kang et al., 2010) before divergent specialized functions developed in different species (Story et al., 2003; Rosenzweig et al., 2005; Cordero-Morales et al., 2011; Geng et al., 2011). TRPA1, like all TRP channels, is expressed as six-transmembrane domain polypeptide subunits, a motif common to many types of ion channels. Functional TRPA1 channels are formed from four of these subunits. Assembled TRPA1 channels are thought to have a homomeric structure (composed of four identical subunits), as there is currently no evidence that heteromultimeric channels involving other TRP channel subunits can form. The channel is distinguished structurally by, and named for, the ∼14–19 ankyrin repeat (AR) domains forming a portion of the protein's intracellular N-terminus. In general, AR domains mediate protein–protein interactions and provide mechanical elasticity (Sedgwick and Smerdon, 1999), although a recent study suggests that particular TRPA1 AR domains can regulate agonist- and heat-induced channel activity (Cordero-Morales et al., 2011). TRPA1 was originally described as a non-selective cation channel that is equally permeable to Na+ versus Ca2+ ions (PCa/PNa reported as 0.84–3.28) (Story et al., 2003; Wang et al., 2008), although Karashima et al. found that during agonist stimulation, PCa/PNa = 7.91 ± 0.60 and the fractional Ca2+ current under these conditions is 17.9–22.3% (Karashima et al., 2010). The unitary conductance of the channel is large (98 pS, when physiological ionic gradients are maintained) (Nagata et al., 2005), indicating that TRPA1 channels can support consequential levels of Ca2+ influx. Predictably, TRPA1 has been shown to influence a broad range of physiological processes that involve Ca2+-dependent signalling pathways, including nociception, mechanotransduction, thermal and oxygen sensing, and responses to environmental irritants and pungent compounds. This manuscript focuses on the role of TRPA1 channels in vascular regulation. The relevant pharmacology is discussed, and studies investigating the consequences of TRPA1 activity on local and integrative control of the vasculature are reviewed.
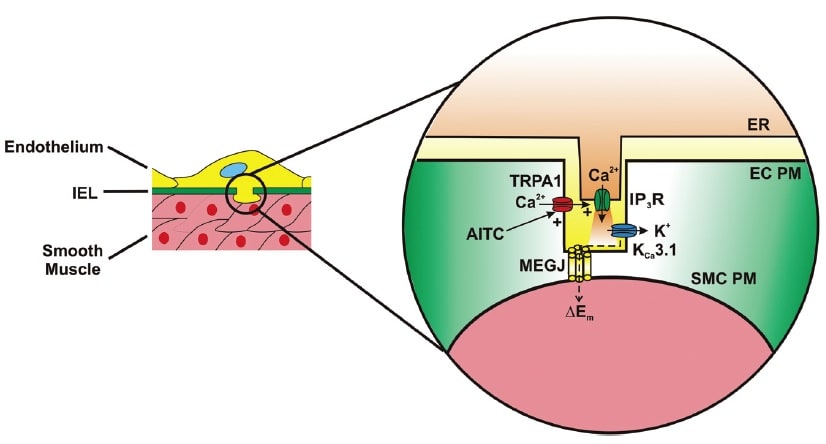
Activation of TRPA1 in cerebral arteries causes endothelium-dependent vasodilation. Allyl isothiocyanate (AITC) activates Ca2+ influx via TRPA1 channels present in myoendothelial junctions in cerebral arteries. TRPA1-mediated Ca2+ influx stimulates Ca2+ release from the endoplasmic reticulum (ER) via inositol trisphosphate receptors (IP3R). The resulting Ca2+ signal (i.e. Ca2+ pulsar) stimulates proximal intermediate conductance Ca2+-activated K+ channels (KCa3.1), resulting in hyperpolarization of the endothelial cell plasma membrane (EC PM). The change in membrane potential (DEm) is conducted via myoendothelial gap junctions (MEGJs) to hyperpolarize the vascular smooth muscle cell plasma membrane (SMCPM), resulting in myocyte relaxation.

Activation of TRPA1 channels in sensory nerves causes arterial dilation. Allyl isothiocyanate (AITC), allicin, cinnamaldehyde (CA) and 4-oxo-2-nonenal (4-ONE) activate Ca2+ influx via TRPA1 channels in sensory nerves, causing release of calcitonin gene-related peptide (CGRP) from perivascular terminals. CGRP binds to its G protein-coupled receptor (GPCR) on the plasma membrane of vascular smooth muscle cells (SMCs) to cause membrane hyperpolarization and myocyte relaxation.
RESEARCH PAPERS
1. メラノコルチン受容体MC1とMC3を介した炎症の保護作用 <炎症のタイムコースが大切>
Chondroprotective and anti-inflammatory role of melanocortin peptides in TNF-α activated human C-20/A4 chondrocytes (pages 67–79)
Magdalena K Kaneva, Mark JP Kerrigan, Paolo Grieco, G Paul Curley, Ian C Locke and Stephen J Getting
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.
BACKGROUND AND PURPOSE
Melanocortin MC1 and MC3 receptors, mediate the anti-inflammatory effects of melanocortin peptides. Targeting these receptors could therefore lead to development of novel anti-inflammatory therapeutic agents. We investigated the expression of MC1 and MC3 receptors on chondrocytes and the role of α-melanocyte-stimulating hormone (α-MSH) and the selective MC3 receptor agonist, [DTRP8]-γ-MSH, in modulating production of inflammatory cytokines, tissue-destructive proteins and induction of apoptotic pathway(s) in the human chondrocytic C-20/A4 cells.
EXPERIMENTAL APPROACH
Effects of α-MSH, [DTRP8]-γ-MSH alone or in the presence of the MC3/4 receptor antagonist, SHU9119, on TNF-α induced release of pro-inflammatory cytokines, MMPs, apoptotic pathway(s) and cell death in C-20/A4 chondrocytes were investigated, along with their effect on the release of the anti-inflammatory cytokine IL-10.
KEY RESULTS
C-20/A4 chondrocytes expressed functionally active MC1,3 receptors. α-MSH and [DTRP8]-γ-MSH treatment, for 30 min before TNF-α stimulation, provided a time-and-bell-shaped concentration-dependent decrease in pro-inflammatory cytokines (IL-1β, IL-6 and IL-8) release and increased release of the chondroprotective and anti-inflammatory cytokine, IL-10, whilst decreasing expression of MMP1, MMP3, MMP13 genes.α-MSH and [DTRP8]-γ-MSH treatment also inhibited TNF-α-induced caspase-3/7 activation and chondrocyte death. The effects of [DTRP8]-γ-MSH, but not α-MSH, were abolished by the MC3/4 receptor antagonist, SHU9119.
CONCLUSION AND IMPLICATIONS
Activation of MC1/MC3 receptors in C-20/A4 chondrocytes down-regulated production of pro-inflammatory cytokines and cartilage-destroying proteinases, inhibited initiation of apoptotic pathways and promoted release of chondroprotective and anti-inflammatory cytokines. Developing small molecule agonists to MC1/MC3 receptors could be a viable approach for developing chondroprotective and anti-inflammatory therapies in rheumatoid and osteoarthritis.
2. PAR1 inhibitor Q94/Q109について
Modulation of PAR1 signalling by benzimidazole compounds (pages 80–94)
S Asteriti, S Daniele, F Porchia, MT Dell' Anno, A Fazzini, I Pugliesi, ML Trincavelli, S Taliani, C Martini, MR Mazzoni and A Gilchrist
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.
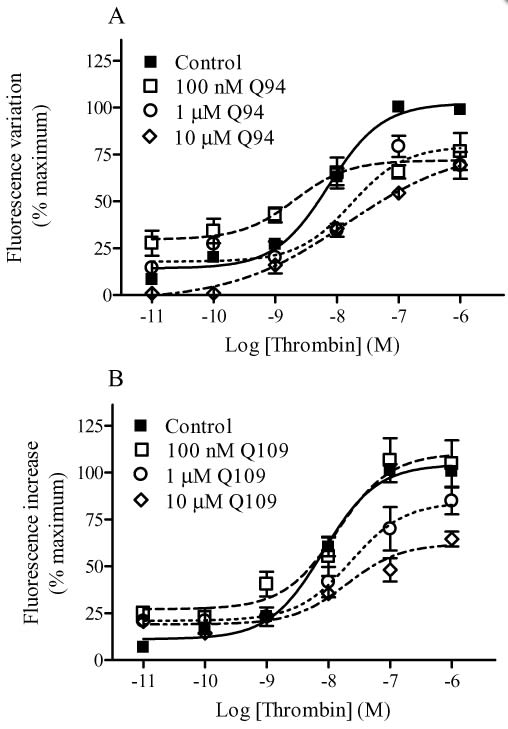
Modulation of thrombin-induced intracellular Ca2+ mobilization by Q94 (A) and Q109 (B). Intracellular Ca2+ mobilization was measured using Fluo 3-AM-loaded HMECs-1 as described in Methods. Basal [Ca2+]i was 240.1 ± 45.3 nM (n = 4). The benzimidazole compounds were added 15 min before the addition of thrombin. The concentration–response curves were performed using seven different concentrations of the enzyme in the presence and absence of fixed concentrations of PAR1 modulators. Data are reported as % of maximal RF and are the means ± SEM of three (A) and four (B) independent experiments, each performed in triplicate.
研究内容:
BACKGROUND AND PURPOSE
Recently, a small molecule (Q94) was reported to selectively block PAR1/Gαq interaction and signalling. Here, we describe the pharmacological properties of Q94 and two analogues that share its benzimidazole scaffold (Q109, Q89). Q109 presents a modest variation from Q94 in the substituent group at the 2-position, while Q89 has quite different groups at the 1- and 2-positions.
EXPERIMENTAL APPROACH
Using human microvascular endothelial cells, we examined intracellular Ca2+ mobilization and inositol 1,4,5-trisphosphate accumulation as well as isoprenaline- or forskolin-stimulated cAMP production in response to thrombin.
KEY RESULTS
Q89 (10 µM) produced a leftward shift in the thrombin-mediated intracellular Ca2+ mobilization concentration–response curve while having no effect on the Emax. Both Q94 (10 µM) and Q109 (10 µM) reduced intracellular Ca2+ mobilization, leading to a decrease in Emax and an increase in EC50 values. Experiments utilizing receptor-specific activating peptides confirmed that Q94 and Q109 were selective for PAR1 as they did not alter the Ca2+ response mediated by a PAR2 activating peptide. Consistent with our Ca2+ results, micromolar concentrations of either Q94 or Q109 significantly reduced thrombin-induced inositol 1,4,5-trisphosphate production. Neither Q94 nor Q109 diminished the inhibitory effects of thrombin on cAMP production, indicating they inhibit signalling selectively through the Gq pathway. Our results also suggest the 1,2-disubstituted benzimidazole derivatives act as ‘allosteric agonists’ of PAR1.
CONCLUSIONS AND IMPLICATIONS
The Q94 and Q109 benzimidazole derivatives represent a novel scaffold for the development of new PAR1 inhibitors and provide a starting point to develop dual signalling pathway-selective positive/negative modulators of PAR1.
Introductionの記載
Protease-activated receptors (PARs) are a family of four GPCRs (PAR1, PAR2, PAR3, and PAR4; receptor nomenclature follows Alexander et al., 2011) characterized by a unique mechanism of activation. PARs are activated enzymatically through proteolysis of the receptor by enzymes of the serine protease family (Macfarlane et al., 2001). The proteolytic cleavage occurs at specific sites within their N-terminal region, thereby exposing novel N-termini, and the ‘tethered ligand’ then folds back onto the extracellular loop II of the receptor, resulting in activation. PAR1, PAR3, and PAR4 are preferentially cleaved by thrombin; whereas PAR2 is mainly a substrate for trypsin, and mast cell tryptase (Coughlin, 2001; Macfarlane et al., 2001; Hollenberg and Compton, 2002). In addition to proteolytic cleavage, most PARs can be activated by synthetic peptides corresponding to the tethered ligand (TL) sequence (Ramachandran and Hollenberg, 2008).
PARs are expressed in many cell types and different organ systems. For example, PAR1, PAR2, PAR3 and PAR4 are all expressed on human endothelial cells (Ramachandran and Hollenberg, 2008); although PAR4 expression may be localized to the endothelium of specific vascular areas (O'Brien et al., 2000; Fujiwara et al., 2005; Hirano et al., 2007). Upon activation, PAR1 exerts its effects on endothelium by activating multiple G-proteins, including Gi/o, Gq/11 and G12/13, leading to modulation of numerous downstream signalling pathways (Barr et al., 1997; Vanhauwe et al., 2002; Ramachandran and Hollenberg, 2008). While PAR1 expression is widely distributed among cells and tissues, PAR2 expression is more limited, and studies indicate that signalling occurs via Gq/11, Gi/o (Nystedt et al., 1995; Macfarlane et al., 2001) and perhaps G12/13 (Ramachandran et al., 2009).
PAR activation plays a key role in many physiological and pathophysiological events involving different organ systems (Ramachandran and Hollenberg, 2008). For example, in the cardiovascular and circulatory systems, activation of PAR1 and to a lesser extent PAR4 on human platelets is sufficient to trigger aggregation (Kahn et al., 1999), while activation of human endothelial PAR1 and PAR2 causes vascular relaxation (Hamilton et al., 2001; 2002; Robin et al., 2003). Indeed, PAR antagonists might prove useful therapeutically for the treatment of several diseases, including thrombosis and atherosclerosis.
Several peptide, peptidomimetic and non-peptide PAR1 antagonists are currently available for experimental studies; and a number of synthetic small molecules are being evaluated for pharmaceutical use in humans (reviewed by Chackalamannil, 2006). In addition, alternative approaches to inhibit PAR1 signalling, such as transfection of endothelial cells with minigene vectors expressing Gα carboxyl (C)-terminal peptides (Gilchrist et al., 2001), or the use of membrane-permeable peptides termed ‘pepducins’ derived from the sequence of the third intracellular loop of PAR1 (Covic et al., 2002a,b), have been presented. Deng et al. (2008) reported the use of a small molecule, Q94, which selectively blocks the interaction between PAR1 and Gαq, to investigate thrombin mediated signalling in mouse lung fibroblasts. Q94 was originally identified during an elisa screen for competition of a high-affinity peptide mimicking the C-terminus of Gαq using a compound library (Deng et al., 2008), and the compound may act as a negative allosteric modulator of PAR1 rather than an orthosteric antagonist. Although Q94 has not been extensively investigated, it represents the first compound to show selective modulation of PAR1/G-protein interactions and thus serve as a biased inhibitor of thrombin-mediated Gq pathway signalling events.
In the present study, we examined the pharmacological properties of Q94 and two analogues, Q109 and Q89, using human microvascular endothelial cells. The three small molecules (Q94, Q109, Q89) all share a benzimidazole scaffold and present either a modest variation in the substituent group at the 2-position (Q109 vs. Q94) or quite different groups at the 1- and 2-positions (Q89 vs. Q94). Whereas micromolar concentrations of Q94 or Q109 resulted in a 30–50% reduction of thrombin's maximal effect (Emax) on intracellular Ca2+ mobilization in combination with a two- to threefold increase of thrombin's EC50 value, a micromolar concentration of Q89 produced a shift to the left in thrombin's concentration–response curve and had no effect on thrombin Emax. Similar to the Ca2+ mobilization studies, experiments assessing inositol-1,4,5-trisphosphate (IP3) accumulation indicated that the presence of micromolar concentrations of either Q94 or Q109 resulted in a significant decrease in thrombin's maximal stimulation (Emax). The antagonistic properties of Q94 and Q109 appear selective as they affected the concentration–response curve of a selective PAR1 activating peptide (AP) while not altering that of a selective PAR2-AP. In addition, these benzimidazole derivatives did not reverse the inhibitory effect of thrombin on isoprenaline- or forskolin-stimulated cAMP production, suggesting they can selectively inhibit the Gq pathway. Importantly, our studies reveal that although the benzimidazole derivatives Q94 and Q109 behave as selective modulators of PAR1 signalling and represent a novel scaffold for the development of new PAR1 inhibitors, their effect on PAR1 signalling is more complex than simple inhibition of Gq activation.
3. IKKβの阻害について
1-Dehydro-[10]-gingerdione from ginger inhibits IKKβ activity for NF-κB activation and suppresses NF-κB-regulated expression of inflammatory genes (pages 128–140)
Hwa Young Lee, Sun Hong Park, Misoon Lee, Hye-Jin Kim, Shi Yong Ryu, Nam Doo Kim, Bang Yeon Hwang, Jin Tae Hong, Sang-Bae Han and Youngsoo Kim
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.
注意事項
Genetic substitutions of the activation loop Ser177 and Ser181 residues with Ala decrease IKKβ activity, whereas those of IKKβ (SS/EE) stimulate kinase activity by mimicking Ser phosphorylation (Mercurio et al., 1997; 1999). Interestingly, IKKβ (C/A) decreases its kinase activity, suggesting that Cys179 plays an important role in the binding affinity of cofactor ATP with IKKβ and in the Ser phosphorylation of IKKβ to stimulate its kinase activity (Byun et al., 2006). To further understand the molecular mechanism of D10G, we proposed molecular docking of D10G to the crystal structure of human IKKβ. The activation loop of IKKβ was somewhat flexible by itself but subtly rearranged to an extended structure upon irreversible binding of D10G with the Cys179 under the most energetically favourable simulation. This conformational change might contribute to the inhibitory mechanism of D10G on IKKβ activity.
BACKGROUND AND PURPOSE
Pungent constituents of ginger (Zingiber officinale) have beneficial effects on inflammatory pain and arthritic swelling. However, the molecular basis for these pharmacological properties is only partially understood. Here, we investigated the molecular target of 1-dehydro-[10]-gingerdione (D10G), one of the pungent constituents of ginger, that mediates its suppression of NF-κB-regulated expression of inflammatory genes linked to toll-like receptor (TLR)-mediated innate immunity.
EXPERIMENTAL APPROACH
RAW 264.7 macrophages or primary macrophages-derived from bone marrows of C57BL/6 or C3H/HeJ mice were stimulated with the TLR4 agonist LPS in the presence of D10G. Catalytic activity of inhibitory κB (IκB) kinase β (IKKβ) was determined by a kinase assay and immunoblot analysis, and the expression of inflammatory genes by RT-PCR analysis and a promoter-dependent reporter assay.
KEY RESULTS
D10G directly inhibited the catalytic activity of cell-free IKKβ. Moreover, D10G irreversibly inhibited cytoplasmic IKKβ-catalysed IκBα phosphorylation in macrophages activated by TLR agonists or TNF-α, and also IKKβ vector-elicited NF-κB transcriptional activity in these cells. These effects of D10G were abolished by substitution of the Cys179 with Ala in the activation loop of IKKβ, indicating a direct interacting site of D10G. This mechanism was shown to mediate D10G-induced disruption of NF-κB activation in LPS-stimulated macrophages and the suppression of NF-κB-regulated gene expression of inducible NOS, COX-2 and IL-6.
CONCLUSION AND IMPLICATIONS
This study demonstrates that IKKβ is a molecular target of D10G involved in the suppression of NF-κB-regulated gene expression in LPS-activated macrophages; this suggests D10G has therapeutic potential in NF-κB-associated inflammation and autoimmune disorders.
彼らのMethods記載
Cell culture
RAW 264.7 macrophages were purchased from ATCC (Manassas, VA). Primary macrophages were prepared from bone marrows of C57BL/6 or C3H/HeJ mice as described previously (Chung et al., 2010). All studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010;McGrath et al., 2010). Animal experiments were carried out following the protocols approved by Animal Experimentation Ethics Committee in CBNU institute. Macrophages were grown in DMEM containing 10% FBS, benzylpenicillin potassium (143 U·mL-1) and streptomycin sulfate (100 µg·mL-1) at 37°C and 5% CO2. RAW 267.4 cells harbuoring the pNF-κB-SEAP-NPT construct were cultured in the same media with an additional supplement of geneticin (500 µg·mL-1).
IKKβ kinase assay
Ser/Thr kinase activity of IKKβ was determined as described previously (Kim et al., 2008). In brief, IKKβ proteins were reacted with substrate GST-IκB (2 µg) and co-factor [γ-32P]-ATP (5 µCi) in a kinase buffer (20 mM HEPES, pH 7.7, 2 mM MgCl2, 50 µM ATP, 10 mM β-glycerophosphate, 10 mM NaF, 300 µg·mL-1 Na3VO4, 2 µM PMSF, 10 µg·mL-1 aprotinin, 1 µg·mL-1 leupeptin, 1 µg·mL-1 pepstatin) at 30°C for 1 h. These reaction mixtures were resolved on SDS-acrylamide gels by electrophoresis. Radioactive bands from the dried gels were then visualized by exposure to X-ray film. Wild-type IKKβ proteins were purchased from Millipore (Billerica, MA, USA). For preparing point-substituted IKKβ proteins, RAW 264.7 cells were transfected with FLAG-tagged expression vector encoding IKKβ (SS/EE) with Glu residues instead of Ser177 and Ser181 or IKKβ (C/A) with Ala residue instead of Cys179 (Kim et al., 2008). Point-substituted IKKβ proteins were precipitated from cell extracts using anti-FLAG affinity gel freezer-safe beads, washed with 20 mM HEPES (pH 7.7) and then subjected to the kinase assay.
4. 鮭カルシトニンでDM治療
Oral salmon calcitonin attenuates hyperglycaemia and preserves pancreatic beta-cell area and function in Zucker diabetic fatty rats (pages 151–163)
M Feigh, KV Andreassen, AV Neutzsky-Wulff, ST Petersen, C Hansen, AC Bay-Jensen, JE Henriksen, H Beck-Nielsen, C Christiansen, K Henriksen and MA Karsdal
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.2012.
BACKGROUND AND PURPOSE
Oral salmon calcitonin (sCT), a dual-action amylin and calcitonin receptor agonist, improved glucose homeostasis in diet-induced obese rats. Here, we have evaluated the anti-diabetic efficacy of oral sCT using parameters of glycaemic control and beta-cell morphology in male Zucker diabetic fatty (ZDF) rats, a model of type 2 diabetes.
EXPERIMENTAL APPROACH
Male ZDF rats were treated with oral sCT (0.5, 1.0 or 2 mg·kg-1) or oral vehicle twice daily from age 8 to 18 weeks. Zucker lean rats served as control group. Fasting and non-fasted blood glucose, glycosylated haemoglobin (HbA1c) and levels of pancreas and incretin hormones were determined. Oral glucose tolerance test and i.p. glucose tolerance test were compared, and beta-cell area and function were evaluated.
KEY RESULTS
Oral sCT treatment dose-dependently attenuated fasting and non-fasted hyperglycaemia during the intervention period. At the end of the study period, oral sCT treatment by dose decreased diabetic hyperglycaemia by ∼9 mM and reduced HbA1c levels by 1.7%. Furthermore, a pronounced reduction in glucose excursions was dose-dependently observed for oral sCT treatment during oral glucose tolerance test. In addition, oral sCT treatment sustained hyperinsulinaemia and attenuated hyperglucagonaemia and hypersecretion of total glucagon-like peptide-1 predominantly in the basal state. Lastly, oral sCT treatment dose-dependently improved pancreatic beta-cell function and beta-cell area at study end.
CONCLUSIONS AND IMPLICATIONS
Oral sCT attenuated diabetic hyperglycaemia in male ZDF rats by improving postprandial glycaemic control, exerting an insulinotropic and glucagonostatic action in the basal state and by preserving pancreatic beta-cell function and beta-cell area.



御礼 誕生日ケーキ・祝賀


最新の画像[もっと見る]
-
 お知らせ 世界敗血症デー 京都 2024
2ヶ月前
お知らせ 世界敗血症デー 京都 2024
2ヶ月前
-
 お知らせ 世界敗血症デー 京都 2024
2ヶ月前
お知らせ 世界敗血症デー 京都 2024
2ヶ月前
-
 お知らせ 世界敗血症デー 京都 2024
2ヶ月前
お知らせ 世界敗血症デー 京都 2024
2ヶ月前
-
 お知らせ 世界敗血症デー 京都 2024
2ヶ月前
お知らせ 世界敗血症デー 京都 2024
2ヶ月前
-
 お知らせ 世界敗血症デー 京都 2024
2ヶ月前
お知らせ 世界敗血症デー 京都 2024
2ヶ月前
-
 セミナーのお知らせ 多発外傷カンファレンス with デューク大学
3ヶ月前
セミナーのお知らせ 多発外傷カンファレンス with デューク大学
3ヶ月前
-
 執筆紹介 麻酔科学と救急医学 ー場の成長と発展のためにー 救急医療 人道の道 JAPAN and the World
3ヶ月前
執筆紹介 麻酔科学と救急医学 ー場の成長と発展のためにー 救急医療 人道の道 JAPAN and the World
3ヶ月前
-
 執筆紹介 麻酔科学と救急医学 ー場の成長と発展のためにー 救急医療 人道の道 JAPAN and the World
3ヶ月前
執筆紹介 麻酔科学と救急医学 ー場の成長と発展のためにー 救急医療 人道の道 JAPAN and the World
3ヶ月前
-
 執筆紹介 麻酔科学と救急医学 ー場の成長と発展のためにー 救急医療 人道の道 JAPAN and the World
3ヶ月前
執筆紹介 麻酔科学と救急医学 ー場の成長と発展のためにー 救急医療 人道の道 JAPAN and the World
3ヶ月前
-
 救急・集中治療 人食いバクテリアの診断と治療
8ヶ月前
救急・集中治療 人食いバクテリアの診断と治療
8ヶ月前
「ジャーナルクラブ 松田直之指導」カテゴリの最新記事
 ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年12月号 Sonic Hed...
ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年12月号 Sonic Hed... 2012年12月13日 基盤ジャーナルクラブ 心血管基礎研究関連
2012年12月13日 基盤ジャーナルクラブ 心血管基礎研究関連 文献紹介 Hypoglycemia and Risk of Death in Critically Ill Patients N Engl J...
文献紹介 Hypoglycemia and Risk of Death in Critically Ill Patients N Engl J... ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年11月号
ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年11月号 ジャーナルクラブ Circulation Res 2012, September 14 & 28
ジャーナルクラブ Circulation Res 2012, September 14 & 28 ジャーナルクラブ British J Pharmacol 2012. October Volume 167, Issue 3
ジャーナルクラブ British J Pharmacol 2012. October Volume 167, Issue 3 ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年9月号
ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年9月号- ジャーナルクラブ Circulation Research 2012. August
 ジャーナルクラブ British Journal of Pharmacology 2012. September
ジャーナルクラブ British Journal of Pharmacology 2012. September ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年7月号
ジャーナルクラブ Am J Physiol Lung Cell Mol Physiol 2012年7月号





