Review
Review
Translational Success Stories: Development of Direct Thrombin Inhibitors
Michiel Coppens, John W. Eikelboom, David Gustafsson, Jeffrey I. Weitz, and Jack Hirsh
Circulation Research. 2012;111:920-929
Anticoagulants are the cornerstone of therapy for conditions associated with arterial and venous thrombosis. Direct thrombin inhibitors (DTIs) are anticoagulants that bind to thrombin and block its enzymatic activity. The bivalent parenteral DTIs hirudin and bivalirudin were based on the observation that the salivary extracts of medicinal leeches prevented blood from clotting. Key events that facilitated the subsequent development of small molecule active site inhibitors, such as argatroban, were the observation that fibrinopeptide A had antithrombotic properties and determination of the crystal structure of thrombin. Hirudin and argatroban have found their niche for the treatment of patients with heparin-induced thrombocytopenia, whereas bivalirudin is approved as an alternative to heparin for patients undergoing percutaneous coronary intervention. The development of orally active direct thrombin inhibitors was challenging because of the need to convert water-soluble, poorly absorbable, active site inhibitors into fat-soluble prodrugs that were then transformed back to the active drug after intestinal absorption. Dabigatran etexilate was the first new oral anticoagulant to be approved for long-term anticoagulant treatment in 6 decades. This Review highlights the development of DTIs as a translational success story; an example in which the combination of scientific ingenuity, structure-based design, and rigorous clinical trials has created a new class of anticoagulants that has improved patient care.

Thrombin has multiple effects on coagulation, platelets, and fibrinolysis and plays a central role in hemostasis. After activation from prothrombin by factor Xa, thrombin acts as a procoagulant protein by cleaving soluble fibrinogen into fibrin monomers, it activates platelets through PAR, and it autocatalytically accelerated its own by activating factor XI and cofactors V and VIII. Once bound to TM, located on the surface of the endothelium, thrombin exerts its anticoagulant and antifibrinolytic effects by activating the natural anticoagulant protein C as well as TAFI. Apart from its functions in hemostasis, thrombin indirectly promotes cell proliferation, migration, and vascular permeability and modulates inflammation through PAR, TAFI, and protein C. Circles with roman numerals represent successively activated clotting factors; squares with roman numerals represent cofactors. Solid arrows represent activation/stimulation; dashed arrows represent inhibition. PAR indicates protease activated receptor; TAFI, thrombin activated fibrinolysis inhibitor; TF, tissue factor; TM, thrombomodulin.
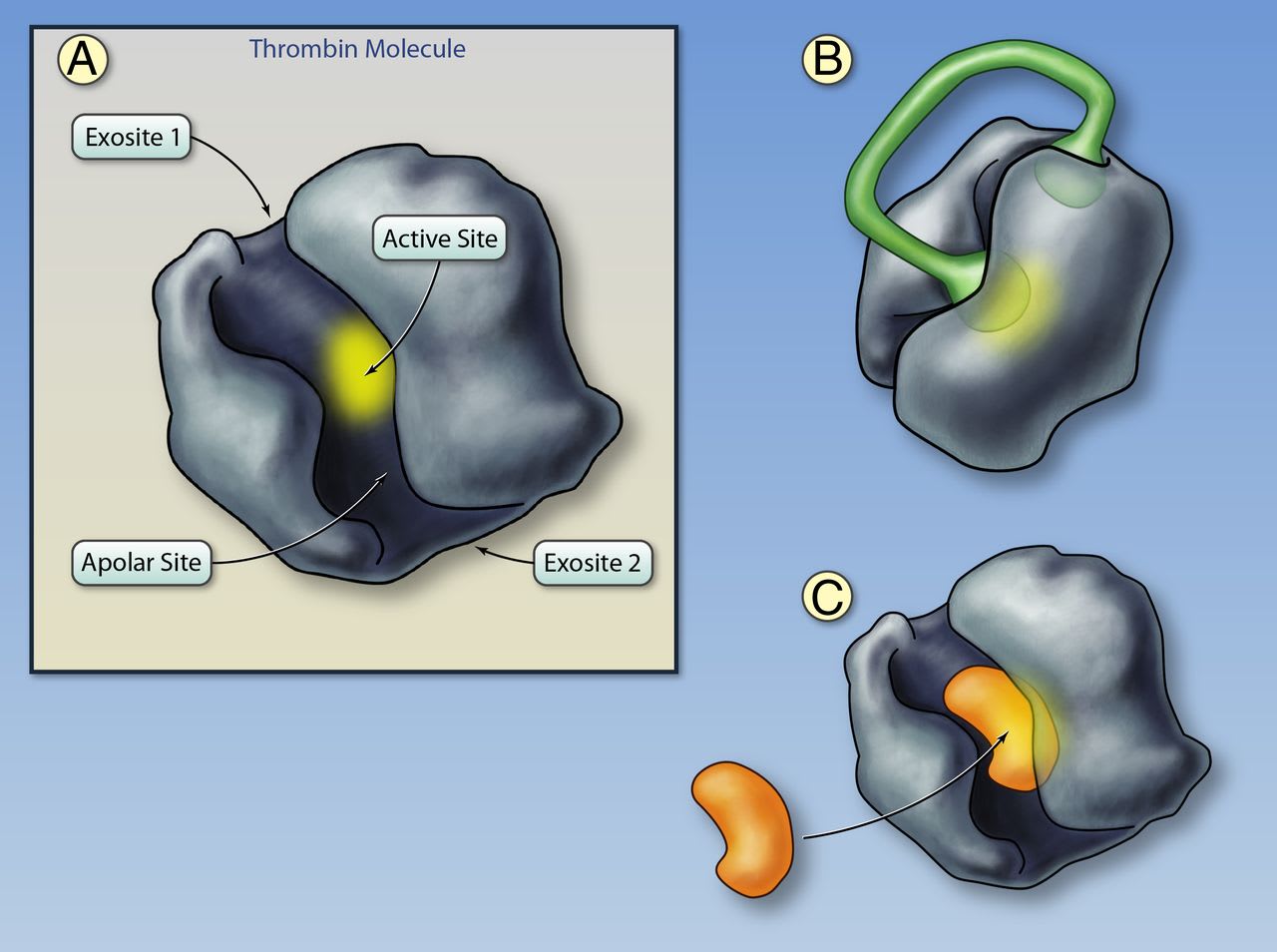
A, Structure of thrombin. The active site of the thrombin molecule is highlighted in yellow; the binding site is located at the base of the cleft. Adjacent to the binding site is the apolar binding site for fibrinogen, fibrinopeptide A, and direct thrombin inhibitors. Exosites I and II are located on opposite sides of the thrombin molecule. Exosite I functions as a substrate docking site, enhancing the affinity of the interaction between thrombin and fibrinogen, protease activated receptors (PARs) on platelets, and cofactors such as thrombomodulin. Exosite II binds to heparin, heparan sulfates, and glycoprotein-Ibα on platelets. B, Thrombin bound to the bivalent direct thrombin inhibitor hirudin. The C-terminus tail of hirudin binds to exosite I and the N-terminus partly obstructs the active site. Binding of bivalirudin is similar with the exception that the N-terminus fully obstructs the active site. After binding, bivalirudin is cleaved by the thrombin active site, which partly restores thrombin activity. C, Thrombin bound to a univalent, small-molecule direct thrombin inhibitor. Univalent direct thrombin inhibitors (argatroban, melagatran, dabigatran) fit into the cleft on thrombin and bind to both the active and apolar sites (Illustration credit: Ben Smith).
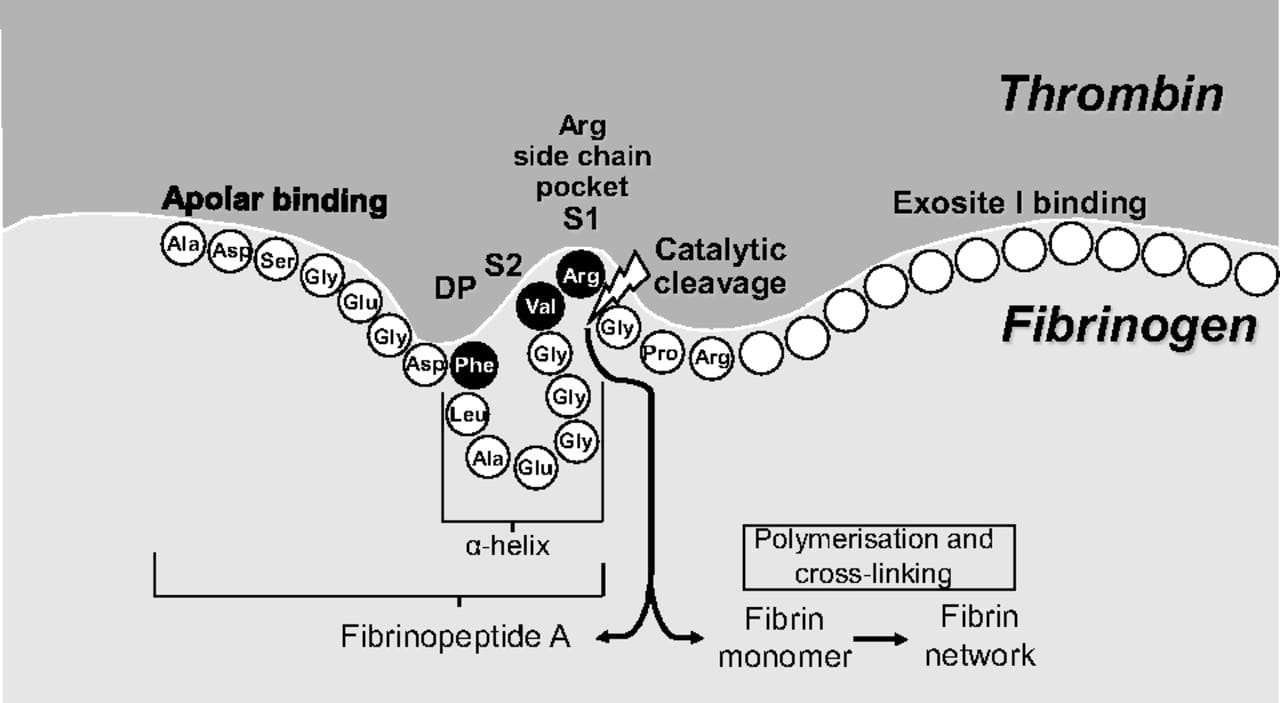
Binding of fibrinogen to the thrombin active site. The α-helix structure of fibrinogen (and fibrinopeptide A) aligns Phe9 with Val2-Arg1, thereby enabling the Phe-Val-Arg tripeptide to bind to the active site of thrombin. This discovery was critical for the development of univalent small-molecule, direct thrombin inhibitors. Arg fits within the Arg side chain pocket (specificity pocket) and cleavage takes place between Arg and the adjacent Gly. S1 and S2 are the pockets for Arg and Val, respectively, and DP is the pocket for Phe. Amino acid abbreviations referred to are Ala, alanine; Arg, arginine; Asp, aspartic acid; Glu, glutamic acid; Gly, glycine; Leu, leucine; Phe, phenylalanine; Pro, proline; Ser, serine; Val, valine (figure adapted with permission from Gustafsson D, et al. Nat Rev Drug Discov. 2004;3:649–659).
Editorial
MicroRNA: A Toolkit Fine-Tuning the Dyadic “Fuzzy Space”?
Long-Sheng Song, Ang Guo, and Richard Z. Lin
Circulation Research. 2012;111:816-818
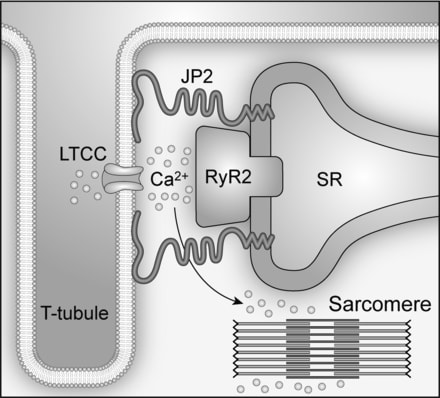
Cardiac excitation-contraction (E-C) coupling links action potentials to muscle contraction and is in essence a process of calcium ion mobilization.1 The central mechanism governing this process in ventricular myocytes is Ca2+-induced Ca2+ release, or CICR. It has been established for more than 20 years that CICR operates in a local control mode, taking place in a restricted junctional space of ≈12 to 15 nm between the transverse (T)-tubule and sarcoplasmic reticulum (SR) membranes, namely, the junctional membrane complexes or cardiac dyads.2,3 Within this dyadic “fuzzy space,”4 clusters of ryanodine receptor (RyR) Ca2+ release channels on the SR constitute the calcium release apparatus together with the directly apposed voltage-gated L-type Ca2+ channels (LTCCs) located primarily on the T-tubule membrane.5 On membrane depolarization, a small amount of Ca2+ influx through the opening of LTCCs locally activates adjacent RyRs to release a much larger (≈10 times) amount of Ca2+ from the SR.6,7 The normal, functional cross-talk between LTCCs and RyRs depends on a stable local ultrastructure―the cardiac dyad.
The molecular mechanism underlying the formation of cardiac dyads remained a mystery until the pioneering work of Takeshima and colleagues.8 In their study, junctophilins were identified as key molecules that maintain junctional membrane complexes between the plasma membrane and the endoplasmic/sarcoplasmic reticulum (ER/SR) in excitable cells. The junctophilin protein family contains 4 members (JP1-4), and JP2 is the only subtype expressed in cardiac myocytes. Lack of JP2 in mice causes embryonic lethality, and JP2 knockout embryonic myocytes have deficient junctional membrane complexes and abnormal Ca2+ signaling, such as reduced intracellular Ca2+ transients.8 Thus, JP2 provides a structural basis for nanoscopic signaling between LTCCs and RyRs during E-C coupling in ventricular myocytes (Figure)
Impaired cardiac E-C coupling/Ca2+ handling is a hallmark of heart failure.9–12 Gomez and colleagues13 first proposed in 1997 that defective E-C coupling is probably due to a change in the relation between RyRs on the SR and LTCCs on T-tubules, although no direct evidence was provided. In the past 10 years, evidence from isolated ventricular myocytes suggests that T-tubule loss and/or disorganization is a significant and common event in advanced heart failure of different etiologies and results in dysynchronous Ca2+ release and impaired contraction.14–23 More recently, the phenomenon of T-tubule remodeling in response to either pressure overload or myocardial infarction was substantiated using an in situ confocal imaging technique in intact hearts.24–26 The reorganization of T-tubule structure alters the spatial organization between LTCCs and RyRs, leading to an increase in orphaned RyRs, the loss of local control of RyRs by LTCCs, Ca2+ release instability, and E-C coupling deficiency in failing myocytes.16,18,21 In addition to T-tubule remodeling, downregulation of JP2 has been found in a variety of heart failure models as well as in failing human hearts.12,24,26–28 Two recent studies in which JP2 was knocked down in either cultured ventricular myocytes24 or by transgenic expression of a JP2 shRNA in mice29 revealed that JP2 downregulation is a key mechanism underlying T-tubule disruption in failing myocytes. The latter study in mice also suggests that JP2 deficiency disrupts the stability of junctional membrane complexes.29 The next logical question is, what is the mechanism responsible for JP2 dysregulation?
In this issue of Circulation Research, Wang and colleagues (Xu et al30) define miR-24 as a novel direct regulator of JP2 homeostasis in the heart. Bioinformatic analysis backed up by experimental data revealed 2 binding sites for miR-24 in the 3′ UTR of JP2 mRNA, either of which was sufficient for maximal repression of JP2 expression. Extending these in vitro studies to models of heart failure demonstrated that miR-24 was upregulated in compensated hypertrophy and in decompensated heart failure, concomitant with loss of JP2 expression and decreased size and volume density of the cardiac dyads. The authors next tested whether overexpression of miR-24 in cultured adult cardiomyocytes could recapitulate the phenotype observed in heart failure model. They found that a 150% increase in miR-24 levels resulted in the anticipated decrease in JP2 expression, which led to a decrease in Ca2+ transient amplitude and E-C coupling gain but no alterations in expression of other E-C coupling proteins. Thus, this study provides novel mechanistic insights into the regulation of JP2 expression in heart cells, adding an important piece to the puzzle of the events that culminate in E-C coupling defects.
The work by Wang and colleagues in this issue of Circulation Research was built on another recent study from the same group (Wu et al31). The objective of the previous study was to understand the ultrastructural mechanism underlying the defective LTCCs-RyRs signaling and compromised contractility in heart failure. Using electron microscopy, the authors found that in response to pressure overload, the size and the volume density of the dyads were significantly reduced. The authors went on to show that knockdown of JP2 replicated the dyadic remodeling observed in the heart failure model, thus suggesting that downregulation of JP2 mechanistically contributes to ultrastructural alterations and loss of E-C coupling in failing hearts. The present study extends these findings to identify miR-24 as a mediator of JP2 downregulation in heart failure.
Over the past few years, miRNAs have gained increasing recognition as important regulators of normal cellular function and disease pathogenesis in many tissues, including the heart.32 An miRNA typically has multiple targets; interestingly, Wang and colleagues (Xu et al30) found that miR-24 overexpression had no impact on other E-C coupling components, and genome-wide scanning did not identify putative miR-24 binding sites in the 3′ UTR of mRNAs encoding other E-C coupling proteins.
The miR-24 binding sites are evolutionally conserved in the 3′ UTR of JP2 mRNAs of mouse, rat, and human origin.30 One important question is whether miR-24 is a physiological regulator of JP2, or if it is only overexpressed in pathological conditions. Cardiac-specific inducible overexpression or knockdown of miR-24 in genetically modified mice should further shed light on this subject. A second question is whether there are other miRNAs that target JP2 for translational silencing. Furthermore, are there posttranslational mechanisms responsible for downregulation of JP2 protein, such as calpain-mediated proteolysis or modifications such as SUMOylation and ubiquitination that target JP2 for degradation? Are there other proteins involved in maintaining the dyadic ultrastructure and T-tubule organization? Answering these questions will further enhance our understanding of cardiac E-C coupling regulation and dysregulation.
Another related question is, how is miR-24 increased in heart failure? Hypertrophic signaling through the calcineurin-NFAT pathway is a well-established mechanism of heart failure.33 Based on a recent report (Lin et al34), Wang and colleagues hypothesized that this pathway induces transcription of the miR-23a/27a/24 cluster as an upstream event in JP2 silencing in heart failure. New studies with genetic modification of the calcineurin-NFAT signaling cascade are necessary to determine whether this pathway indeed mediates the increase of miR-24 and subsequent JP2 loss.
An important future experiment will be to examine whether introduction of an antago-miR against miR-24 protects against development or progression of heart failure. These data would not only confirm the role of miR-24 in JP2 downregulation in heart failure but would also indicate whether antago-miR should be pursued as a novel therapeutic strategy to augment JP2 expression in the treatment of heart failure.
原著
1. ヘパリンによるCXCR4ブロック作用の危険性 Heparin Disrupts the CXCR4/SDF-1 Axis and Impairs the Functional Capacity of Bone Marrow–Derived Mononuclear Cells Used for Cardiovascular Repair
Florian H. Seeger, Tina Rasper, Ariane Fischer, Marion Muhly-Reinholz, Eduard Hergenreider, David M. Leistner, Katharina Sommer, Yosif Manavski, Reinhard Henschler, Emmanouil Chavakis, Birgit Assmus, Andreas M. Zeiher, and Stefanie Dimmeler
Circulation Research. 2012;111:854-862
Abstract
Rationale: Cell therapy is a promising option for the treatment of acute or chronic myocardial ischemia. The intracoronary infusion of cells imposes the potential risk of cell clotting, which may be prevented by the addition of anticoagulants. However, a comprehensive analysis of the effects of anticoagulants on the function of the cells is missing.
Objective: Here, we investigated the effects of heparin and the thrombin inhibitor bivalirudin on bone marrow–derived mononuclear cell (BMC) functional activity and homing capacity.
Methods and Results: Heparin, but not bivalirudin profoundly and dose-dependently inhibited basal and stromal cell–derived factor 1 (SDF-1)–induced BMC migration. Incubation of BMCs with 20 U/mL heparin for 30 minutes abrogated SDF-1–induced BMC invasion (16±8% of control; P<0.01), whereas no effects on apoptosis or colony formation were observed (80±33% and 100±44% of control, respectively). Pretreatment of BMCs with heparin significantly reduced the homing of the injected cells in a mouse ear-wound model (69±10% of control; P<0.05). In contrast, bivalirudin did not inhibit in vivo homing of BMCs. Mechanistically, heparin binds to both, the chemoattractant SDF-1 and its receptor, chemokine receptor 4 (CXCR4), blocking CXCR4 internalization as well as SDF-1/CXCR4 signaling after SDF-1 stimulation.
Conclusions: Heparin blocks SDF-1/CXCR4 signaling by binding to the ligand as well as the receptor, thereby interfering with migration and homing of BMCs. In contrast, the thrombin inhibitor bivalirudin did not interfere with BMC homing or SDF-1/CXCR4 signaling. These findings suggest that bivalirudin but not heparin might be recommended as an anticoagulant for intracoronary infusion of BMCs for cell therapy after cardiac ischemia.
<img src="http://blogimg.goo.ne.jp/user_image/05/ff/4f10622a72f6316124e2f85e8ec3ce90.png" border="0">
Proposed mechanism. Under normal conditions, stromal cell–derived factor-1 (SDF-1) binds to its receptor, chemokine receptor 4 (CXCR4). The complex internalizes and initiates downstream signaling. Heparin can bind to the chemokine SDF-1, building a heparin–SDF-1 complex. Heparin-bound SDF-1 demonstrates reduced CXCR4 binding ability and thereby impaired downstream signaling. Moreover, heparin can bind directly to the CXCR4 receptor. SDF-1 can still bind to the CXCR4 heparin complex; however, this complex demonstrates reduced internalization and impaired downstream signaling after SDF-1 stimulation.
 Reviews
Reviews
Oral Direct Factor Xa Inhibitors
Calvin H. Yeh, James C. Fredenburgh, and Jeffrey I. Weitz
Circulation Research. 2012;111:1069-1078
Vitamin K antagonists, such as warfarin, have been the mainstay of oral anticoagulation for many decades. Although effective, warfarin has numerous limitations, including a variable dose requirement from patient to patient because of differences in dietary vitamin K intake, common genetic polymorphisms, and multiple drug interactions that affect its pharmacodynamics and metabolism. Consequently, warfarin requires frequent monitoring to ensure that a therapeutic anticoagulant effect has been achieved because excessive anticoagulation can lead to bleeding, and because insufficient anticoagulation can result in thrombosis. Such monitoring is burdensome for patients and physicians and is costly for the health care system. These limitations have prompted the development of new oral anticoagulants that target either factor Xa or thrombin. Although the path to the development of these drugs has been long, the new drugs are at least as effective and safe as warfarin, but they streamline clinical care because they can be administered in fixed doses without routine coagulation monitoring. This article focuses on rivaroxaban, apixaban, and edoxaban, the oral factor Xa inhibitors in the most advanced stages of development. After 20 years of discovery research, these agents are already licensed for several indications. Thus, the long path to finding replacements for warfarin has finally reached fruition. Therefore, development of the oral factor Xa inhibitors represents a translational science success story.

Critical role of factor Xa in coagulation. The extrinsic pathway is initiated when tissue factor (TF) exposed at sites of vascular injury binds activated factor VII (fVIIa) on the activated membrane surface to form extrinsic tenase (Xase). The extrinsic tenase complex activates factors IX and X. Activated factor IX (fIXa) binds to activated factor VIII (fVIIIa) on the activated membrane surface to form the intrinsic tenase complex. IXa also may be generated via the contact pathway after vascular injury. Both the extrinsic and intrinsic tenase complexes generate activated factor X (fXa), which assembles with activated factor V (fVa) on the activated membrane surface to form prothrombinase. The resulting thrombin then converts fibrinogen to fibrin and activates platelets, resulting in the formation of a platelet-fibrin thrombus. By targeting the active site of fXa, the oral fXa inhibitors attenuate thrombin generation that is triggered either via extrinsic tenase or via intrinsic tenase.
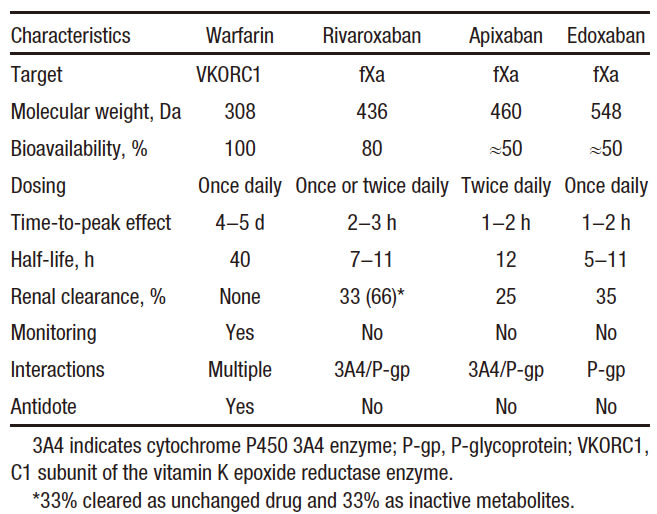
Pharmacological Properties of Warfarin, Rivaroxaban, Apixaban, and Edoxaban
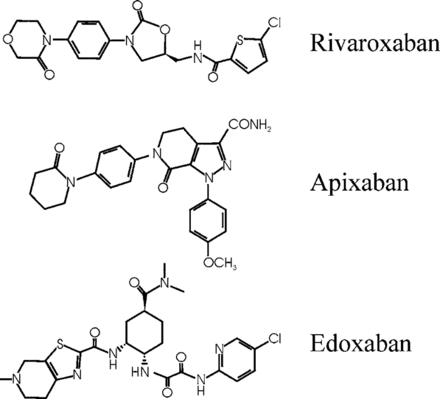
Structures of rivaroxaban, apixaban, and edoxaban, the oral factor Xa inhibitors, in the most advanced stages of development.

Landmark Phase III Clinical Trials and the Number of Patients Enrolled for Rivaroxaban, Apixaban, and Edoxaban for Various Indications
Editorials
G Protein–Coupled Receptor Kinase 5: Exploring Its Hype in Cardiac Hypertrophy
Stephen L. Belmonte and Burns C. Blaxall
Circulation Research. 2012;111:957-958
It is an established dogma that G-protein–coupled receptor kinases (GRKs) classically direct the desensitization and internalization of their eponymous receptors through direct phosphorylation. Yet, it is the noncanonical action of GRKs that has increasingly attracted the interest of groups seeking novel insight into vexing pathophysiological questions. Among the 7 GRK isoforms, several findings suggest that GRK5 may have particular relevance to the development of cardiac hypertrophy and heart failure (HF). Beyond its classical function, GRK5 contains a DNA-binding nuclear localization sequence1 and has recently been reported to modify myocardial gene transcription through histone deacetylase kinase activity.2 Furthermore, GRK5 expression is elevated in the ventricles of patients with HF,3 and transgenic cardiac-specific GRK5 overexpression produced pronounced hypertrophy and accelerated HF progression upon pressure-overload challenge in mice.2
To date, the question remains whether endogenous GRK5 is a prerequisite for hypertrophy and HF development in the face of cardiac stress. In this issue of Circulation Research, Gold et al4 have begun to address this issue by using global and cardiac-restricted GRK5 knockout mice. They report that when subjected to transverse aortic constriction, both the global and cardiac-restricted GRK5 null animals show delayed hypertrophy and preserved heart function compared with control mice, as determined by serial left ventricular posterior wall thickness assessment and ejection fraction, respectively. Furthermore, they used semiquantitative polymerase chain reaction to demonstrate that mRNA expression of a host of hypertrophy marker genes was significantly reduced in the hearts of both GRK5 knockout mice. Similar results were obtained after chronic administration of a subpressor dose of phenylephrine in the global GRK5 knockout mice. Overall, the data suggest that GRK5 is integral to maladaptive cardiac hypertrophy.
In evaluating the results presented in the article by Gold et al,4 it becomes apparent that GRK5 may represent a viable therapeutic target for the treatment of HF, for which new therapies are desperately needed. As with most new and exciting discoveries, there are various facets of GRK5 biology to consider before clinical application. For example, it was previously reported that the global GRK5 knockout mouse exhibits enhanced muscarinic receptor sensitivity but no gross anatomic differences from wild-type littermates.5 In addition, a recent study found high expression of GRK5 in white adipose tissue, underlying reduced adipogenesis and obesity in GRK5-null animals.6 Thus, while cardiac functional parameters clearly suggest a protective role of GRK5 in the heart, it would also be valuable to assess ratios of heart weight to tibia length in addition to body weight, as well as to quantify myocyte size. It is interesting to note that both global and cardiac-restricted GRK5 knockout mice demonstrated progressive, mild (possibly compensatory) cardiac hypertrophy after transverse aortic constriction, whereas the wild-type mice followed the more traditional progression of concentric hypertrophy followed by rapidly decompensated, eccentric hypertrophy coupled to ventricular wall thinning. Attenuation of the hypertrophic gene expression profile after myocardial insult in GRK5 knockouts, partially explained by modest but significant alterations in non-nuclear histone deacetylase phosphorylation, further validates an important role for GRK5 in pathological cardiac hypertrophy.
Importantly, GRK5 is expressed in multiple cardiac cell types. In their current article, Gold et al4 report relatively similar results in both the global and cardiomyocyte-restricted GRK5 mice. Future investigation will be needed to determine the possible functional relevance of GRK5 in the maladaptive hypertrophic response in various nonmyocyte cardiac cells (eg, fibroblasts). Interestingly, prior studies have documented divergent effects of altered GRK5 expression/activity. For example, hybrid transgenic mice overexpressing cardiac GRK5 and a constitutively active α1B-adrenergic receptor mutant demonstrated that GRK5 reduced α1B-adrenergic receptor hypertrophy and partially reduced atrial natriuretic factor mRNA.7 Although somewhat inconsistent with the current report, the inherent differences in the mechanism of injury induced by an activated mutant receptor, transverse aortic constriction, and persistent agonist stimulation may, in large part, explain the discrepancy. It is known that GRK5 phosphorylates and desensitizes α1B-adrenergic receptor basally but not after agonist stimulation.8 Furthermore, the α1B-adrenergic receptor is preferentially expressed in cardiac fibroblasts, whereas the α1A subtype predominates in myocytes.9 It is also likely that GRK5 overexpression confers protection via enhanced desensitization of β-adrenergic receptors, as observed previously in mice.10 This is also the putative explanation for the improved outcomes of patients with HF with a highly active GRK5 polymorphism.11 Considering that GRK5 exhibits differential receptor subtype specificity and has both nuclear and membrane receptor kinase activity, further work is required to establish a definitive role for GRK5 in maladaptive cardiac hypertrophy in a variety of cardiac cell types.
One final point to address is how GRK5 regulates gene transcription. Hypertrophic gene expression is primarily considered in terms of upregulated genes, but downregulated genes are also clinically relevant to hypertrophy and HF. Indeed, it was recently reported that in mice overexpressing Gαq, which produces a pressure-overload cardiac phenotype, enhanced GRK5 expression normalized a subset of downregulated mRNAs responsible for carbohydrate metabolism and energy production.12 Furthermore, expression of a truncated GRK5 that expresses the regulator of G protein signaling homology domain, which inhibits nuclear factor-κB transcriptional activity, reduced left ventricular hypertrophy in spontaneously hypertensive rats or normotensive rats exposed to chronic phenelyephrine.13 Taken together, these results suggest that GRK5 expression counteracts at least some types of hypertrophic stimuli.
In summary, GRK5 expression is clearly pertinent to maladaptive cardiac hypertrophy and the development of HF. Further advances in our understanding of the functional role of GRK5 in heart failure, including those reported by Gold et al,4 may serve to clarify some inconsistencies and take the quest for a new HF therapeutic yet one step closer to reality.









 アルテルナリア
アルテルナリア