Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia of the Lung (DIPNECH): Current Best Evidence.
Wirtschafter E1, Walts AE, Liu ST, Marchevsky AM.
Lung. 2015 Jun 24.
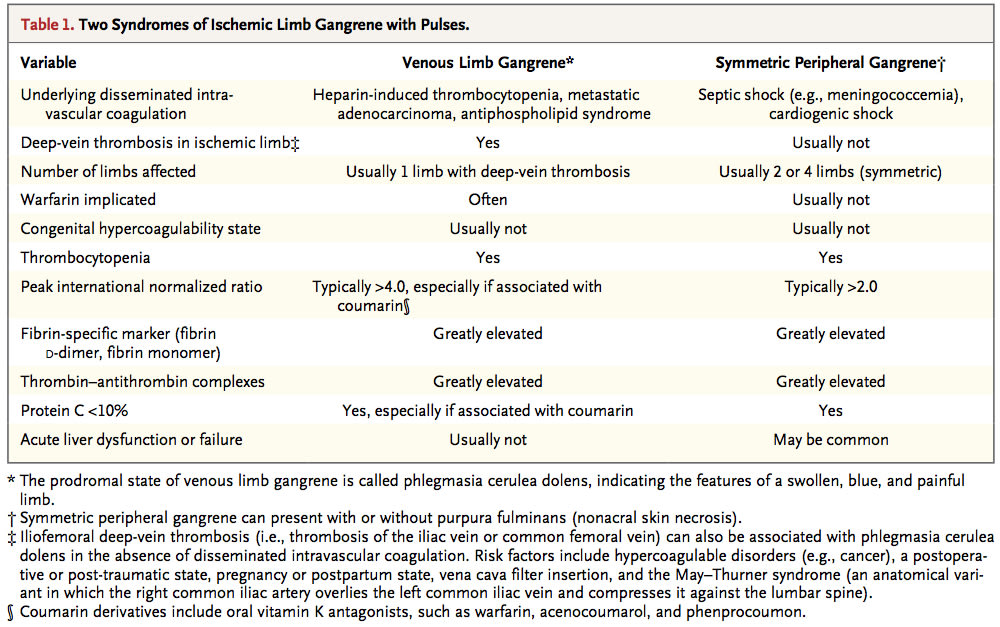
Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) is recognized as a preneoplastic condition by the World Health Organization. We reviewed our experience with 30 patients and performed a systematic review of the English literature to collect best evidence on the clinical features and disease course in 169 additional patients. Some patients presented with one or more carcinoid tumors associated with multiple small pulmonary nodules on imaging studies and showed DIPNECH as a somewhat unexpected pathologic finding. Others presented with multiple small pulmonary nodules that raised suspicion of metastatic disease on imaging. A third subset was presented with previously unexplained respiratory symptoms. In most patients, DIPNECH was associated with a good prognosis, with chronological progression into a subsequent carcinoid tumor noted in only one patient and death attributed directly to DIPNECH in only two patients. There is no best evidence to support the use of octreotide, steroids, or bronchodilators in DIPNECH patients.
http://pubs.rsna.org/doi/full/10.1148/rg.336135506
DIPNECH: when to suggest this diagnosis on CT.
Chassagnon G, Favelle O, Marchand-Adam S, De Muret A, Revel MP.
Clin Radiol. 2015;70:317-25.
(2014) Sarcoid-Like Reaction in Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia. American Journal of Respiratory and Critical Care Medicine 190:10, e62-e63
(2012) mTOR/p70S6K in Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia. American Journal of Respiratory and Critical Care Medicine 185:3, 341-342
(2011) Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia:A Systematic Overview
Am J Respir Crit Care Med184:8–16
※ 当科の管理するoncological ICUの肺傷害の管理として,念頭に置いています。
Wirtschafter E1, Walts AE, Liu ST, Marchevsky AM.
Lung. 2015 Jun 24.
Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) is recognized as a preneoplastic condition by the World Health Organization. We reviewed our experience with 30 patients and performed a systematic review of the English literature to collect best evidence on the clinical features and disease course in 169 additional patients. Some patients presented with one or more carcinoid tumors associated with multiple small pulmonary nodules on imaging studies and showed DIPNECH as a somewhat unexpected pathologic finding. Others presented with multiple small pulmonary nodules that raised suspicion of metastatic disease on imaging. A third subset was presented with previously unexplained respiratory symptoms. In most patients, DIPNECH was associated with a good prognosis, with chronological progression into a subsequent carcinoid tumor noted in only one patient and death attributed directly to DIPNECH in only two patients. There is no best evidence to support the use of octreotide, steroids, or bronchodilators in DIPNECH patients.
http://pubs.rsna.org/doi/full/10.1148/rg.336135506
DIPNECH: when to suggest this diagnosis on CT.
Chassagnon G, Favelle O, Marchand-Adam S, De Muret A, Revel MP.
Clin Radiol. 2015;70:317-25.
(2014) Sarcoid-Like Reaction in Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia. American Journal of Respiratory and Critical Care Medicine 190:10, e62-e63
(2012) mTOR/p70S6K in Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia. American Journal of Respiratory and Critical Care Medicine 185:3, 341-342
(2011) Diffuse Idiopathic Pulmonary Neuroendocrine Cell Hyperplasia:A Systematic Overview
Am J Respir Crit Care Med184:8–16
※ 当科の管理するoncological ICUの肺傷害の管理として,念頭に置いています。