








特発性肺線維症におけるSonic Hedgehogシグナルの関与
Role of Sonic Hedgehog in idiopathic pulmonary fibrosis.
Bolaños AL, Milla CM, Lira JC, Ramírez R, Checa M, Barrera L, García-Alvarez J, Carbajal V, Becerril C, Gaxiola M, Pardo A, Selman M.
Am J Physiol Lung Cell Mol Physiol. 2012 Dec;303(11):L978-90.
The Hedgehog System Machinery Controls Transforming Growth Factor-β-Dependent Myofibroblastic Differentiation in Humans: Involvement in Idiopathic Pulmonary Fibrosis.
Cigna N, Farrokhi Moshai E, Brayer S, Marchal-Somme J, Wémeau-Stervinou L, Fabre A, Mal H, Lesèche G, Dehoux M, Soler P, Crestani B, Mailleux AA.
Am J Pathol. 2012 Dec;181(6):2126-37.
MicroRNAs in idiopathic pulmonary fibrosis.
Pandit KV, Milosevic J, Kaminski N.
Transl Res. 2011 Apr;157(4):191-9. doi: 10.1016/j.trsl.2011.01.012. Epub 2011 Feb 4. Review.
参考 RT-PCR
Forward Reverse
SHH 5′-GAAAGCAGAGAACTCGGTGG-3′ 5′-CTCAGGTCCTTCACCAGCTT-3′
IHH 5′-TCAGCCTGCTCTCACTACGA-3′ 5′-CAAAGGGGCCTAAGATGGAT-3′
DHH 5′-GACCGCAACAAGTATGGGTT-3′ 5′-TATCAGCTTTGACCGACACG-3′
PTC 5′-TGGCATAGGAGTGGAGTTCA-3′ 5′-ATCAGCACTCCCAGCAGAGT-3′
HHIP 5′-CATGTCGTCATGGAGGTGTC-3′ 5′-GCGGATGTTTCTGTCCACTT-3′
SMO 5′-CAGCAAGATCAACGAGACCA-3′ 5′-GGCAGCTGAAGGTAATGAGC-3′
GLI1 5′-CCAGCCAGAGAGACCAACA-3′ 5′-ATCCGACAGAGGTGAGATGG-3′
GLI2 5′-TTTATGGGCATCCTCTCTGG-3′ 5′-AAGGCTGGAAAGCACTGTGT-3′
GLI3 5′-CCTCCCAACTCCTCACACAT-3′ 5′-CAACACCAACTGGTCCCTCT-3′
SUFU 5′-ACATGCTGCTGACAGAGGAC-3′ 5′-CAGACACCAACGATCTGGAG-3′
SPOP 5′-TTCTGCGAGGTGAGTGTTGT-3′ 5′-GGCACTCAGGAACCTTTACC-3′
UBC 5′-CACTTGGTCCTGCGCTTGA-3′ 5′-TTTTTTGGGAATGCAACAACTT-3′
αSMA 5′-GAAGAGCATCCCACCCTGC-3′ 5′-ATTTTCTCCCGGTTGGCCT-3′
FN1 5′-GCCAACAGGATGACATGAAT-3′ 5′-CATACCCGCCGAATGTAGGA-3′
α2COL-1 5′-TTGAGACTCAGCCACCCAGAGT-3′ 5′-CAGTTCTTGGCTGGGATGTTTT-3′
Hedgehog pathwayのIPF関連分子mRNA levelの修飾作用
Immunostaining Positive regulators
SHH ↓ Positive in fibrotic lesions; ↓ in “normal” areas
IHH ↓ ND
DHH = ND
SMO = =
GLI1 = Nuclear in fibrotic lesions
GLI2 = Nuclear in fibrotic lesions and in “normal” areas
SUFU ↓ Nuclear in fibrotic lesions
Negative regulators
PTC ↓ ↓ in fibrotic lesions
HHIP ↓ ND
GLI3 = ↓ in fibrotic lesions
SPOP ↓ ND
↓, decreased compared with control lung tissue; =, unchanged; ND, not determined.


1. 総説 Neuronal Norepinephrine Transporter (NET)の異常について
C. Schroeder and J. Jordan
Norepinephrine transporter function and human cardiovascular disease
Am J Physiol Heart Circ Physiol December 1, 2012 303:H1273-H1282
pproximately 80–90% of the norepinephrine released in the brain or in peripheral tissues is taken up again through the neuronal norepinephrine transporter (NET). Pharmacological studies with NET inhibitors showed that NET has opposing effects on cardiovascular sympathetic regulation in the brain and in the periphery. Furthermore, NET is involved in the distribution of sympathetic activity between vasculature, heart, and kidney. Genetic NET dysfunction is a rare cause of the postural tachycardia syndrome. The condition is characterized by excessive adrenergic stimulation of the heart, particularly with standing. Conversely, NET inhibition may be beneficial in hypoadrenergic states, such as central autonomic failure or neurally mediated syncope, which results from acute sympathetic withdrawal. Biochemical studies suggested reduced NET function in some patients with essential hypertension. Furthermore, cardiac NET function appears to be reduced in common heart diseases, such as congestive heart failure, ischemic heart disease, and stress-induced cardiomyopathy. Whether NET dysfunction is a consequence or cause of progressive heart disease in human subjects requires further study. However, studies with the nonselective NET inhibitor sibutramine suggest that reduced NET function could have an adverse effect on the cardiovascular system. Given the widespread use of medications inhibiting NET, the issue deserves more attention.
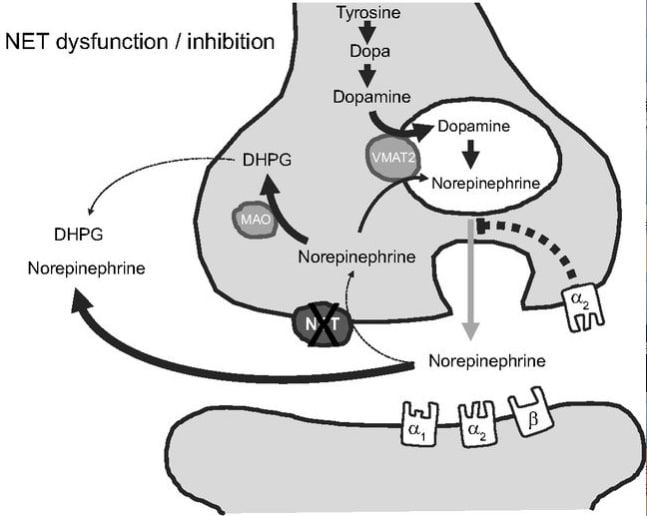
Schematic diagram of norepinephrine biosynthesis, release, reuptake, and degradation. For details see text. DHPG, dihydroxyphenylglycol; NET, norepinephrine reuptake transporter; MAO, monoaminooxidase; VMAT2, vesicular monoamine transporter-2.
2. 総説 ミトコンドリアNOSについて<虚血再潅流研究の知識として重要>
Tamara Zaobornyj and Pedram Ghafourifar
Strategic localization of heart mitochondrial NOS: a review of the evidence
Am J Physiol Heart Circ Physiol December 1, 2012 303:H1283-H129
Heart mitochondria play a central role in cell energy provision and in signaling. Nitric oxide (NO) is a free radical with primary regulatory functions in the heart and involved in a broad array of key processes in cardiac metabolism. Specific NO synthase (NOS) isoforms are confined to distinct locations in cardiomyocytes. The present article reviews the chemical reactions through which NO interacts with biomolecules and exerts some of its crucial roles. Specifically, the article discusses the reactions of NO with mitochondrial targets and the subcellular localization of NOS within the myocardium and analyzes the available data about heart mitochondrial NOS activity and identity. The article also describes the regulation of heart mtNOS by the distinctive mitochondrial environment by showing the effects of Ca2+, O2, L-arginine, mitochondrial transmembrane potential, and the metabolic states on heart mitochondrial NO production. The article depicts the effects of NO on heart function and highlights the relevance of NO production within mitochondria. Finally, the evidence on the functional implications of heart mitochondrial NOS is delineated with emphasis on chronic hypoxia and ischemia-reperfusion studies.
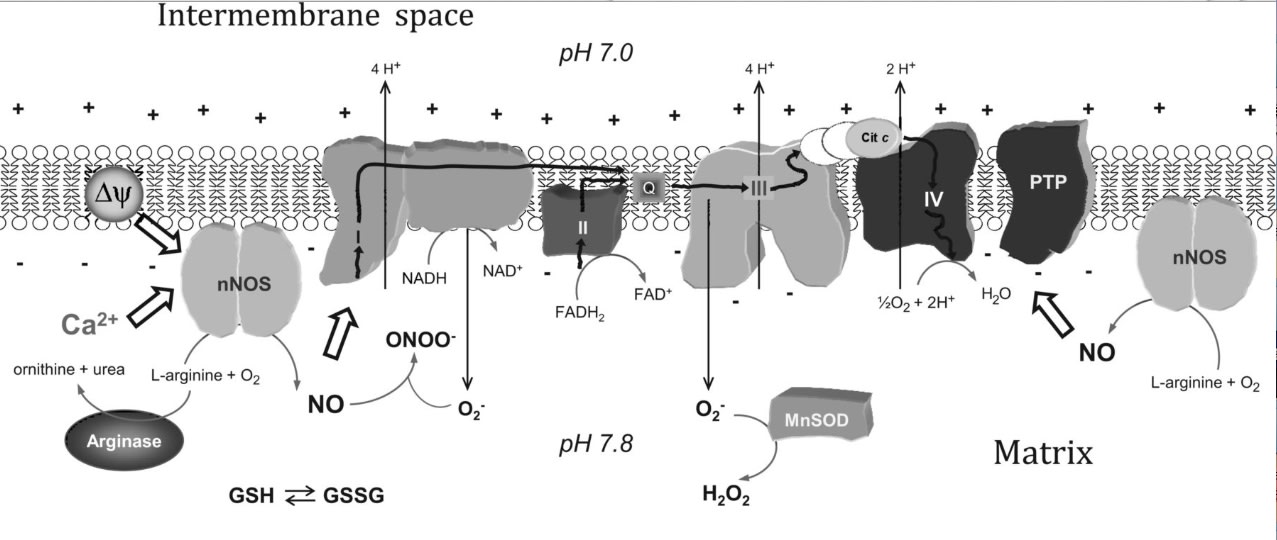
Reciprocal regulation of mtNOS activity and mitochondrial function. Localization of NOS within mitochondria provides a distinct specific regulation of mitochondrial NO production by intramitochondrial O2, Ca2+, pH, mitochondrial transmembrane potential (Δψ), L-arginine, arginases, or redox state (GSH/GSGG balance). NO produced by mtNOS can readily react with mitochondrial targets such as respiratory complexes I, III, or IV or mitochondrial PTP. The superoxide anion (O2-) is formed at the respiratory chain and undergoes a very fast reaction with NO to form peroxynitrite (ONOO-), or it is catabolized by MnSOD to form H2O2. Cit c, cytochrome c.
3. 総説 Circulation miRNAについて
Anke J. Tijsen, Yigal M. Pinto, and Esther E. Creemers
Circulating microRNAs as diagnostic biomarkers for cardiovascular diseases
Am J Physiol Heart Circ Physiol November 1, 2012 303:H1085-H1095
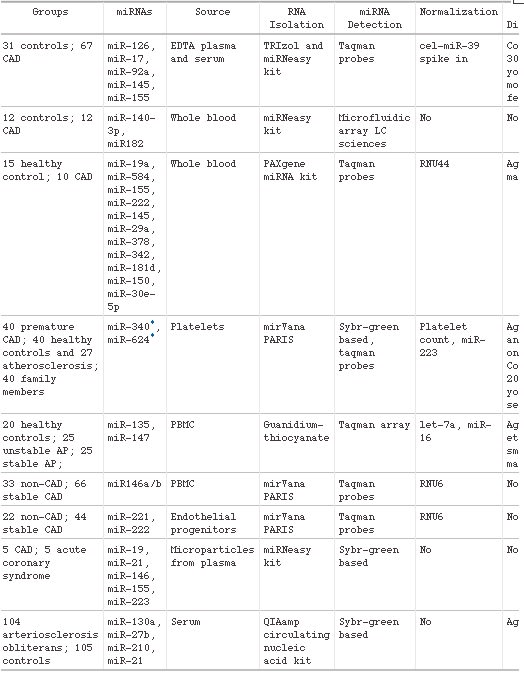
One of the major challenges in cardiovascular disease is the identification of reliable clinical biomarkers that can be routinely measured in plasma. MicroRNAs (miRNAs) were recently discovered to circulate in the bloodstream in a remarkably stable form. Because of their stability and often tissue- and disease-specific expression and the possibility to measure them with high sensitivity and specificity, miRNAs are emerging as new diagnostic biomarkers. In this review we will provide an overview of the potential of circulating miRNAs as biomarkers for a wide range of cardiovascular diseases such as coronary artery disease, myocardial infarction, hypertension, heart failure, viral myocarditis, and type-2 diabetes mellitus. Furthermore, we will discuss the challenges with regard to further validation in large patient cohorts, and we will discuss how the measurement of multiple miRNAs simultaneously might improve the accuracy of the diagnostic test.
Circulating miRNAs as biomarkers for CAD
4. miR-10のFlt-1の発現調節作用
MicroRNAs Control Vascular Endothelial Growth Factor Signaling
Reinier A. Boon
Circ Res 2012 Nov 9;111(11):1388-90

Angiogenesis is a very tightly controlled process, in which endothelial cells need to migrate and proliferate toward ischemic tissue. A long-known factor that provides a gradient for endothelial cells to migrate toward is vascular endothelial growth factor (VEGF).1 Carefully titrated levels of VEGF are crucial for blood vessel development, because even the heterozygous deletion of VEGF is lethal in mice.2 In the past decades, it has become clear that VEGF signaling is very complex, with different VEGF isoforms and VEGF receptors that tightly control endothelial cell behavior. Interestingly, although VEGF receptor 2 (kinase insert domain receptor [KDR]) is one of the main proangiogenic VEGF receptors,3 binding of VEGF to VEGF receptor 1 (fms-related tyrosine kinase 1 [FLT1]) does not result in proangiogenic signaling, which raised the concept that FLT1 acts as a trap or decoy for VEGF. Thus, FLT1 can negatively regulate VEGF signaling, and this is of crucial importance, for instance, to keep the cornea avascular,4 but also aids in controlling the fine balance between proangiogenic and antiangiogenic factors. To make matters more complex, FLT1 mRNA is alternatively spliced, giving rise to a membrane-bound FLT1 and secreted soluble FLT1,5 but they both function as VEGF traps, preventing VEGF from activating KDR. In this issue of Circulation Research, Hassel et al6 report that both isoforms of FLT1 are regulated by the microRNA (miRNA) miR-10.
Hypoglycemia and Risk of Death in Critically Ill Patients
NICE-SUGAR Study Investigators, Finfer S, Liu B, Chittock DR, Norton R, Myburgh JA, McArthur C, Mitchell I, Foster D, Dhingra V, Henderson WR, Ronco JJ, Bellomo R, Cook D, McDonald E, Dodek P, Hébert PC, Heyland DK, Robinson BG.
N Engl J Med 2012;367:1108-18.
低血糖により死亡率が高まること,集中治療管理における低血糖発症のリスク因子がまとめられています。
Abstract
BACKGROUND:
Whether hypoglycemia leads to death in critically ill patients is unclear.
METHODS:
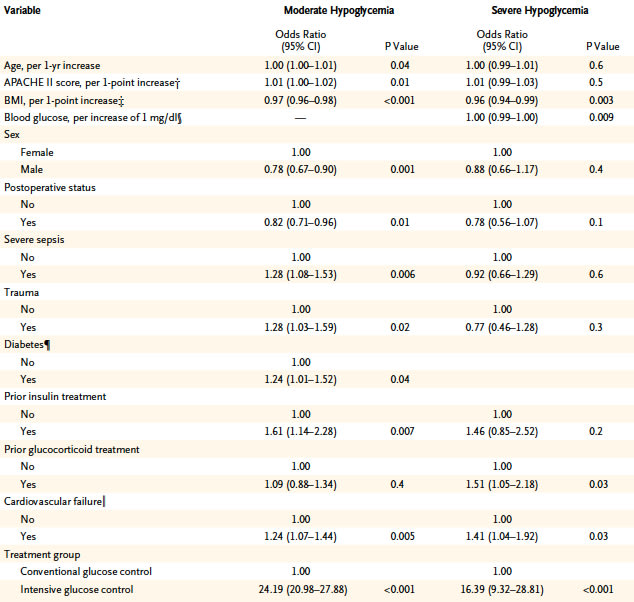
We examined the associations between moderate and severe hypoglycemia (blood glucose, 41 to 70 mg per deciliter [2.3 to 3.9 mmol per liter] and ≤40 mg per deciliter [2.2 mmol per liter], respectively) and death among 6026 critically ill patients in intensive care units (ICUs). Patients were randomly assigned to intensive or conventional glucose control. We used Cox regression analysis with adjustment for treatment assignment and for baseline and postrandomization covariates.
RESULTS:
Follow-up data were available for 6026 patients: 2714 (45.0%) had moderate hypoglycemia, 2237 of whom (82.4%) were in the intensive-control group (i.e., 74.2% of the 3013 patients in the group), and 223 patients (3.7%) had severe hypoglycemia, 208 of whom (93.3%) were in the intensive-control group (i.e., 6.9% of the patients in this group). Of the 3089 patients who did not have hypoglycemia, 726 (23.5%) died, as compared with 774 of the 2714 with moderate hypoglycemia (28.5%) and 79 of the 223 with severe hypoglycemia (35.4%). The adjusted hazard ratios for death among patients with moderate or severe hypoglycemia, as compared with those without hypoglycemia, were 1.41 (95% confidence interval [CI], 1.21 to 1.62; P1 day vs. 1 day, P=0.01), those who died from distributive (vasodilated) shock (P CONCLUSIONS:
In critically ill patients, intensive glucose control leads to moderate and severe hypoglycemia, both of which are associated with an increased risk of death. The association exhibits a dose-response relationship and is strongest for death from distributive shock. However, these data cannot prove a causal relationship.
1.TLR4 KOは肺胞型上皮細胞のオートファジーを促進させる
Chang Hyeok An, Xiao Mei Wang, Hilaire C. Lam, Emeka Ifedigbo, George R. Washko, Stefan W. Ryter, and Augustine M. K. Choi
TLR4 deficiency promotes autophagy during cigarette smoke-induced pulmonary emphysema
Am J Physiol Lung Cell Mol Physiol;2012 303:L748-L757
TLR4 KOマウスやTLR4 siRNAを用いて,肺胞2型上皮細胞を解析すると,正常状態と比較してTLR4 KOマウスやTLR4 siRNAで,LC3およびcaspase3の活性が上昇しているという。肺胞2型上皮細胞からの分泌リガンドの抑制,FADDの上昇に対する効果,抗アポトーシスファクター産生の抑制が関与するのであろうが,詳細の評価が不十分な内容である。現象論としての記載に留まっている。
2.肺動脈平滑筋におけるgap junctionalを介したTGF-βシグナル
Salina Gairhe, Natalie N. Bauer, Sarah A. Gebb, and Ivan F. McMurtry
Serotonin passes through myoendothelial gap junctions to promote pulmonary arterial smooth muscle cell differentiation
Am J Physiol Lung Cell Mol Physiol; 2012 303:L767-L777
Myoendothelial gap junctional signaling mediates pulmonary arterial endothelial cell (PAEC)-induced activation of latent TGF-β and differentiation of cocultured pulmonary arterial smooth muscle cells (PASMCs), but the nature of the signal passing from PAECs to PASMCs through the gap junctions is unknown. Because PAECs but not PASMCs synthesize serotonin, and serotonin can pass through gap junctions, we hypothesized that the monoamine is the intercellular signal. We aimed to determine whether PAEC-derived serotonin mediates PAEC-induced myoendothelial gap junction-dependent activation of TGF-β signaling and differentiation of PASMCs. Rat PAECs and PASMCs were monocultured or cocultured with (touch) or without (no-touch) direct cell-cell contact. In all cases, tryptophan hydroxylase 1 (Tph1) transcripts were expressed predominantly in PAECs. Serotonin was detected by immunostaining in both PAECs and PASMCs in PAEC/PASMC touch coculture but was not found in PASMCs in either PAEC/PASMC no-touch coculture or in PASMC/PASMC touch coculture. Furthermore, inhibition of gap junctions but not of the serotonin transporter in PAEC/PASMC touch coculture prevented serotonin transfer from PAECs to PASMCs. Inhibition of serotonin synthesis pharmacologically or by small interfering RNAs to Tph1 in PAECs inhibited the PAEC-induced activation of TGF-β signaling and differentiation of PASMCs. We concluded that serotonin synthesized by PAECs is transferred through myoendothelial gap junctions into PASMCs, where it activates TGF-β signaling and induces a more differentiated phenotype. This finding suggests a novel role of gap junction-mediated intercellular serotonin signaling
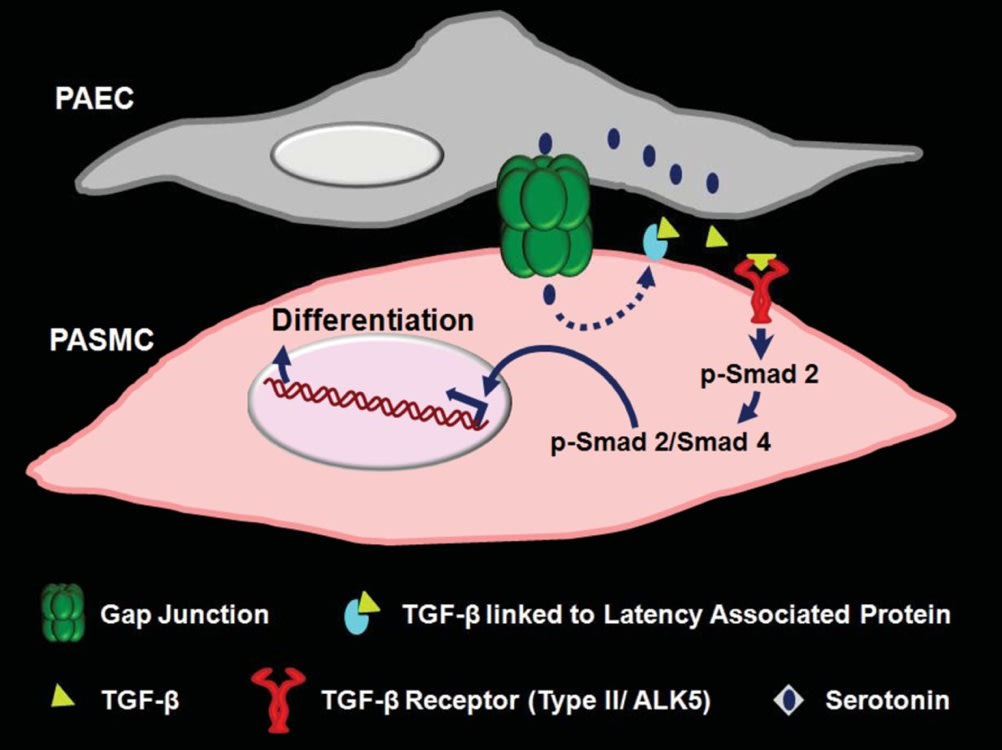
Model for PAEC-induced, myoendothelial gap junction-dependent, serotonin-mediated differentiation of cocultured PASMCs. PAEC and PASMC cocultured with direct cell-cell contact mimic the in vivo environment by forming myoendothelial gap junctions. Serotonin synthesized in PAECs passes directly into PASMCs through myoendothelial gap junctions and, by an unknown mechanism, activates latent TGF-β by dissociating active TGF-β from latency-associated protein. Activated TGF-β via its receptor ALK5 then triggers a signaling cascade that phosphorylates SMAD 2. p-SMAD 2 and SMAD 4 interact, and the complex translocates to the nucleus, where it stimulates the synthesis of smooth muscle cell differentiation marker proteins (contractile proteins such as MHC, H1-calponin, and α-SMC .
3.CSE(cigarette smoke extract)がユビキチンープロテアソーム系を傷害する
Sabine H. van Rijt, Ilona E. Keller, Gerrit John, Kathrin Kohse, Ali Ö. Yildirim, Oliver Eickelberg, and Silke Meiners
Acute cigarette smoke exposure impairs proteasome function in the lung
Am J Physiol Lung Cell Mol Physiol November 1, 2012 303:L814-L823
Cigarette smoke mediates DNA damage, lipid peroxidation, and modification and misfolding of proteins, thereby inducing severe cellular damage. The ubiquitin proteasome system serves as the major disposal system for modified and misfolded proteins and is thus essential for proper cellular function. Its role in cigarette smoke-induced cell damage, however, is largely unknown. We hypothesized that the ubiquitin-proteasome system is involved in the degradation of cigarette smoke-damaged proteins and that cigarette smoke exposure impairs the proteasome itself. Here, we show that treatment of human alveolar epithelial cells with cigarette smoke extract (CSE) induced time- and dose-dependent cell death, a rise in intracellular reactive oxygen species, and increased levels of carbonylated and polyubiquitinated proteins. While high doses of CSE severely impaired all three proteasomal activities, low CSE concentrations significantly inhibited only the trypsin-like activity of the proteasome in alveolar and bronchial epithelial cells. Moreover, acute exposure of mice to cigarette smoke significantly impaired the trypsin-like activity by 25% in the lungs. Reduced proteasome activity was not due to transcriptional regulation of the proteasome. Notably, cigarette smoke exposure induced accumulation of polyubiquitinated proteins in the soluble and insoluble protein fraction of the lung. We show for the first time that acute exposure to cigarette smoke directly impairs proteasome activity in the lungs of mice and in human epithelial cells at low doses without affecting proteasome expression. Our results indicate that defective proteasomal protein quality control may exacerbate the detrimental effects of cigarette smoke in the lung.

Review
Translational Success Stories: Development of Direct Thrombin Inhibitors
Michiel Coppens, John W. Eikelboom, David Gustafsson, Jeffrey I. Weitz, and Jack Hirsh
Circulation Research. 2012;111:920-929
Anticoagulants are the cornerstone of therapy for conditions associated with arterial and venous thrombosis. Direct thrombin inhibitors (DTIs) are anticoagulants that bind to thrombin and block its enzymatic activity. The bivalent parenteral DTIs hirudin and bivalirudin were based on the observation that the salivary extracts of medicinal leeches prevented blood from clotting. Key events that facilitated the subsequent development of small molecule active site inhibitors, such as argatroban, were the observation that fibrinopeptide A had antithrombotic properties and determination of the crystal structure of thrombin. Hirudin and argatroban have found their niche for the treatment of patients with heparin-induced thrombocytopenia, whereas bivalirudin is approved as an alternative to heparin for patients undergoing percutaneous coronary intervention. The development of orally active direct thrombin inhibitors was challenging because of the need to convert water-soluble, poorly absorbable, active site inhibitors into fat-soluble prodrugs that were then transformed back to the active drug after intestinal absorption. Dabigatran etexilate was the first new oral anticoagulant to be approved for long-term anticoagulant treatment in 6 decades. This Review highlights the development of DTIs as a translational success story; an example in which the combination of scientific ingenuity, structure-based design, and rigorous clinical trials has created a new class of anticoagulants that has improved patient care.

Thrombin has multiple effects on coagulation, platelets, and fibrinolysis and plays a central role in hemostasis. After activation from prothrombin by factor Xa, thrombin acts as a procoagulant protein by cleaving soluble fibrinogen into fibrin monomers, it activates platelets through PAR, and it autocatalytically accelerated its own by activating factor XI and cofactors V and VIII. Once bound to TM, located on the surface of the endothelium, thrombin exerts its anticoagulant and antifibrinolytic effects by activating the natural anticoagulant protein C as well as TAFI. Apart from its functions in hemostasis, thrombin indirectly promotes cell proliferation, migration, and vascular permeability and modulates inflammation through PAR, TAFI, and protein C. Circles with roman numerals represent successively activated clotting factors; squares with roman numerals represent cofactors. Solid arrows represent activation/stimulation; dashed arrows represent inhibition. PAR indicates protease activated receptor; TAFI, thrombin activated fibrinolysis inhibitor; TF, tissue factor; TM, thrombomodulin.
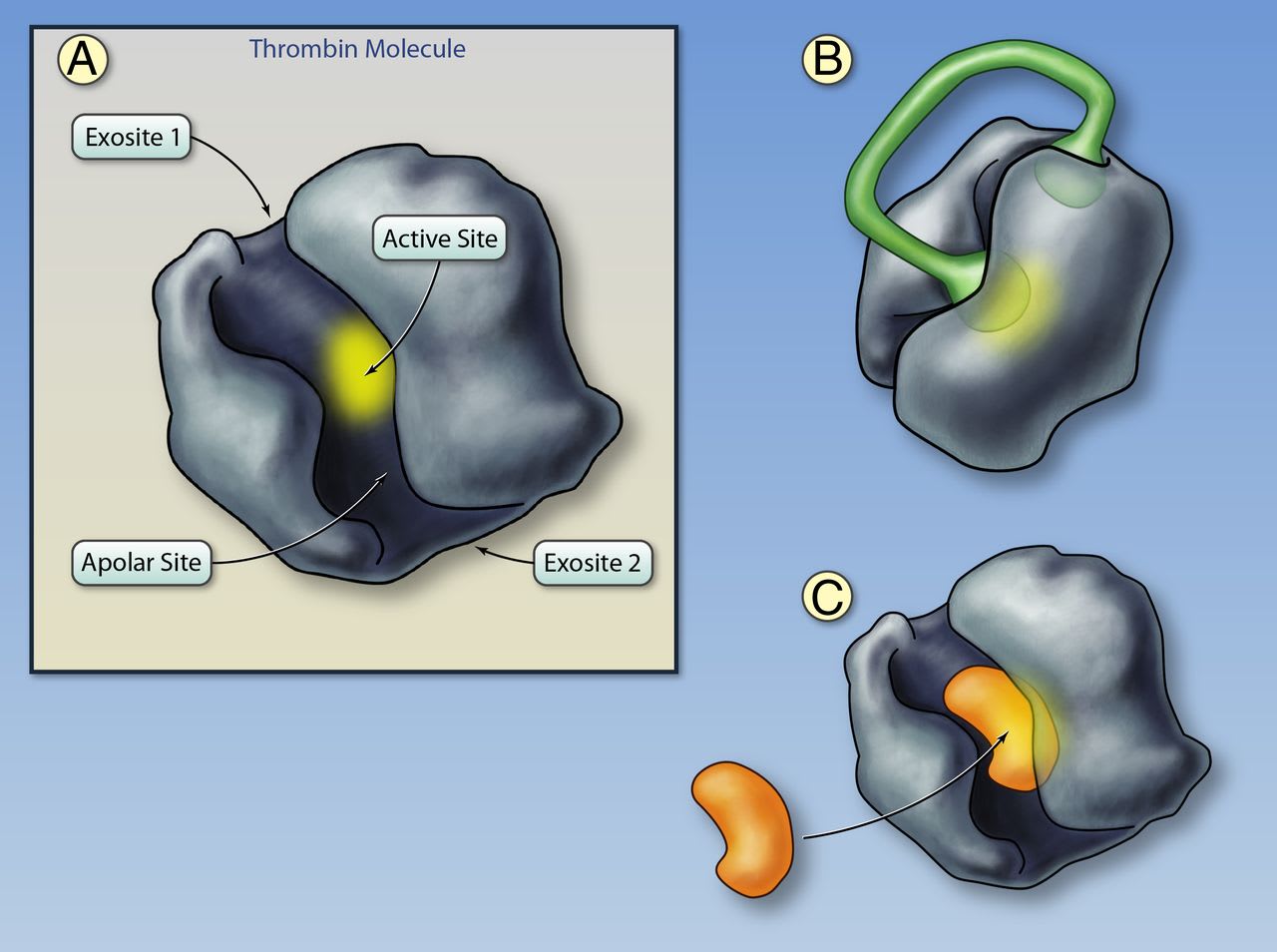
A, Structure of thrombin. The active site of the thrombin molecule is highlighted in yellow; the binding site is located at the base of the cleft. Adjacent to the binding site is the apolar binding site for fibrinogen, fibrinopeptide A, and direct thrombin inhibitors. Exosites I and II are located on opposite sides of the thrombin molecule. Exosite I functions as a substrate docking site, enhancing the affinity of the interaction between thrombin and fibrinogen, protease activated receptors (PARs) on platelets, and cofactors such as thrombomodulin. Exosite II binds to heparin, heparan sulfates, and glycoprotein-Ibα on platelets. B, Thrombin bound to the bivalent direct thrombin inhibitor hirudin. The C-terminus tail of hirudin binds to exosite I and the N-terminus partly obstructs the active site. Binding of bivalirudin is similar with the exception that the N-terminus fully obstructs the active site. After binding, bivalirudin is cleaved by the thrombin active site, which partly restores thrombin activity. C, Thrombin bound to a univalent, small-molecule direct thrombin inhibitor. Univalent direct thrombin inhibitors (argatroban, melagatran, dabigatran) fit into the cleft on thrombin and bind to both the active and apolar sites (Illustration credit: Ben Smith).
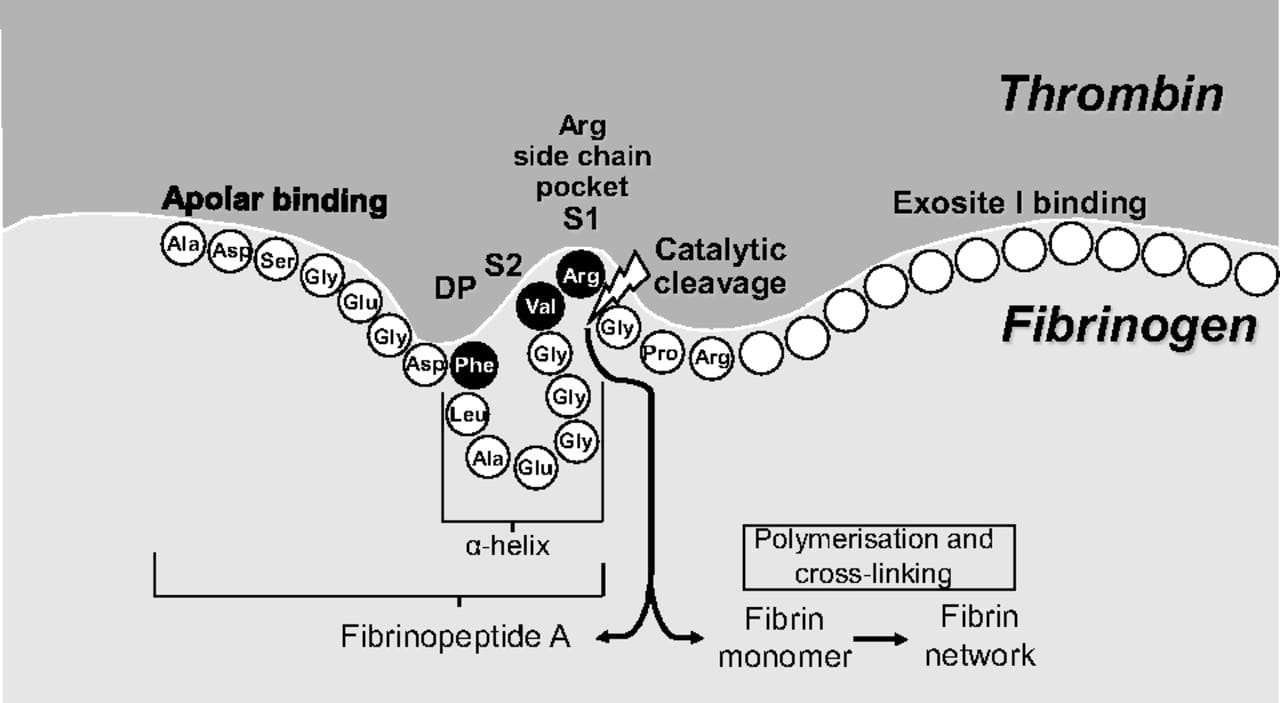
Binding of fibrinogen to the thrombin active site. The α-helix structure of fibrinogen (and fibrinopeptide A) aligns Phe9 with Val2-Arg1, thereby enabling the Phe-Val-Arg tripeptide to bind to the active site of thrombin. This discovery was critical for the development of univalent small-molecule, direct thrombin inhibitors. Arg fits within the Arg side chain pocket (specificity pocket) and cleavage takes place between Arg and the adjacent Gly. S1 and S2 are the pockets for Arg and Val, respectively, and DP is the pocket for Phe. Amino acid abbreviations referred to are Ala, alanine; Arg, arginine; Asp, aspartic acid; Glu, glutamic acid; Gly, glycine; Leu, leucine; Phe, phenylalanine; Pro, proline; Ser, serine; Val, valine (figure adapted with permission from Gustafsson D, et al. Nat Rev Drug Discov. 2004;3:649–659).
Editorial
MicroRNA: A Toolkit Fine-Tuning the Dyadic “Fuzzy Space”?
Long-Sheng Song, Ang Guo, and Richard Z. Lin
Circulation Research. 2012;111:816-818
Cardiac excitation-contraction (E-C) coupling links action potentials to muscle contraction and is in essence a process of calcium ion mobilization.1 The central mechanism governing this process in ventricular myocytes is Ca2+-induced Ca2+ release, or CICR. It has been established for more than 20 years that CICR operates in a local control mode, taking place in a restricted junctional space of ≈12 to 15 nm between the transverse (T)-tubule and sarcoplasmic reticulum (SR) membranes, namely, the junctional membrane complexes or cardiac dyads.2,3 Within this dyadic “fuzzy space,”4 clusters of ryanodine receptor (RyR) Ca2+ release channels on the SR constitute the calcium release apparatus together with the directly apposed voltage-gated L-type Ca2+ channels (LTCCs) located primarily on the T-tubule membrane.5 On membrane depolarization, a small amount of Ca2+ influx through the opening of LTCCs locally activates adjacent RyRs to release a much larger (≈10 times) amount of Ca2+ from the SR.6,7 The normal, functional cross-talk between LTCCs and RyRs depends on a stable local ultrastructure―the cardiac dyad.
The molecular mechanism underlying the formation of cardiac dyads remained a mystery until the pioneering work of Takeshima and colleagues.8 In their study, junctophilins were identified as key molecules that maintain junctional membrane complexes between the plasma membrane and the endoplasmic/sarcoplasmic reticulum (ER/SR) in excitable cells. The junctophilin protein family contains 4 members (JP1-4), and JP2 is the only subtype expressed in cardiac myocytes. Lack of JP2 in mice causes embryonic lethality, and JP2 knockout embryonic myocytes have deficient junctional membrane complexes and abnormal Ca2+ signaling, such as reduced intracellular Ca2+ transients.8 Thus, JP2 provides a structural basis for nanoscopic signaling between LTCCs and RyRs during E-C coupling in ventricular myocytes (Figure)
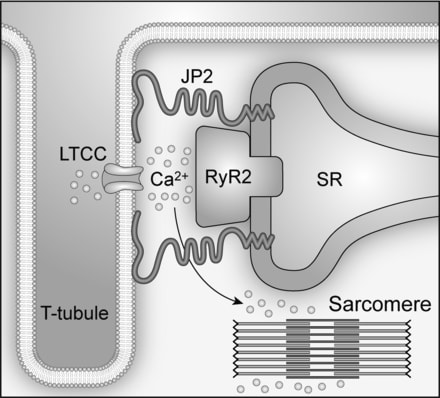
Impaired cardiac E-C coupling/Ca2+ handling is a hallmark of heart failure.9–12 Gomez and colleagues13 first proposed in 1997 that defective E-C coupling is probably due to a change in the relation between RyRs on the SR and LTCCs on T-tubules, although no direct evidence was provided. In the past 10 years, evidence from isolated ventricular myocytes suggests that T-tubule loss and/or disorganization is a significant and common event in advanced heart failure of different etiologies and results in dysynchronous Ca2+ release and impaired contraction.14–23 More recently, the phenomenon of T-tubule remodeling in response to either pressure overload or myocardial infarction was substantiated using an in situ confocal imaging technique in intact hearts.24–26 The reorganization of T-tubule structure alters the spatial organization between LTCCs and RyRs, leading to an increase in orphaned RyRs, the loss of local control of RyRs by LTCCs, Ca2+ release instability, and E-C coupling deficiency in failing myocytes.16,18,21 In addition to T-tubule remodeling, downregulation of JP2 has been found in a variety of heart failure models as well as in failing human hearts.12,24,26–28 Two recent studies in which JP2 was knocked down in either cultured ventricular myocytes24 or by transgenic expression of a JP2 shRNA in mice29 revealed that JP2 downregulation is a key mechanism underlying T-tubule disruption in failing myocytes. The latter study in mice also suggests that JP2 deficiency disrupts the stability of junctional membrane complexes.29 The next logical question is, what is the mechanism responsible for JP2 dysregulation?
In this issue of Circulation Research, Wang and colleagues (Xu et al30) define miR-24 as a novel direct regulator of JP2 homeostasis in the heart. Bioinformatic analysis backed up by experimental data revealed 2 binding sites for miR-24 in the 3′ UTR of JP2 mRNA, either of which was sufficient for maximal repression of JP2 expression. Extending these in vitro studies to models of heart failure demonstrated that miR-24 was upregulated in compensated hypertrophy and in decompensated heart failure, concomitant with loss of JP2 expression and decreased size and volume density of the cardiac dyads. The authors next tested whether overexpression of miR-24 in cultured adult cardiomyocytes could recapitulate the phenotype observed in heart failure model. They found that a 150% increase in miR-24 levels resulted in the anticipated decrease in JP2 expression, which led to a decrease in Ca2+ transient amplitude and E-C coupling gain but no alterations in expression of other E-C coupling proteins. Thus, this study provides novel mechanistic insights into the regulation of JP2 expression in heart cells, adding an important piece to the puzzle of the events that culminate in E-C coupling defects.
The work by Wang and colleagues in this issue of Circulation Research was built on another recent study from the same group (Wu et al31). The objective of the previous study was to understand the ultrastructural mechanism underlying the defective LTCCs-RyRs signaling and compromised contractility in heart failure. Using electron microscopy, the authors found that in response to pressure overload, the size and the volume density of the dyads were significantly reduced. The authors went on to show that knockdown of JP2 replicated the dyadic remodeling observed in the heart failure model, thus suggesting that downregulation of JP2 mechanistically contributes to ultrastructural alterations and loss of E-C coupling in failing hearts. The present study extends these findings to identify miR-24 as a mediator of JP2 downregulation in heart failure.
Over the past few years, miRNAs have gained increasing recognition as important regulators of normal cellular function and disease pathogenesis in many tissues, including the heart.32 An miRNA typically has multiple targets; interestingly, Wang and colleagues (Xu et al30) found that miR-24 overexpression had no impact on other E-C coupling components, and genome-wide scanning did not identify putative miR-24 binding sites in the 3′ UTR of mRNAs encoding other E-C coupling proteins.
The miR-24 binding sites are evolutionally conserved in the 3′ UTR of JP2 mRNAs of mouse, rat, and human origin.30 One important question is whether miR-24 is a physiological regulator of JP2, or if it is only overexpressed in pathological conditions. Cardiac-specific inducible overexpression or knockdown of miR-24 in genetically modified mice should further shed light on this subject. A second question is whether there are other miRNAs that target JP2 for translational silencing. Furthermore, are there posttranslational mechanisms responsible for downregulation of JP2 protein, such as calpain-mediated proteolysis or modifications such as SUMOylation and ubiquitination that target JP2 for degradation? Are there other proteins involved in maintaining the dyadic ultrastructure and T-tubule organization? Answering these questions will further enhance our understanding of cardiac E-C coupling regulation and dysregulation.
Another related question is, how is miR-24 increased in heart failure? Hypertrophic signaling through the calcineurin-NFAT pathway is a well-established mechanism of heart failure.33 Based on a recent report (Lin et al34), Wang and colleagues hypothesized that this pathway induces transcription of the miR-23a/27a/24 cluster as an upstream event in JP2 silencing in heart failure. New studies with genetic modification of the calcineurin-NFAT signaling cascade are necessary to determine whether this pathway indeed mediates the increase of miR-24 and subsequent JP2 loss.
An important future experiment will be to examine whether introduction of an antago-miR against miR-24 protects against development or progression of heart failure. These data would not only confirm the role of miR-24 in JP2 downregulation in heart failure but would also indicate whether antago-miR should be pursued as a novel therapeutic strategy to augment JP2 expression in the treatment of heart failure.
原著
1. ヘパリンによるCXCR4ブロック作用の危険性 Heparin Disrupts the CXCR4/SDF-1 Axis and Impairs the Functional Capacity of Bone Marrow–Derived Mononuclear Cells Used for Cardiovascular Repair
Florian H. Seeger, Tina Rasper, Ariane Fischer, Marion Muhly-Reinholz, Eduard Hergenreider, David M. Leistner, Katharina Sommer, Yosif Manavski, Reinhard Henschler, Emmanouil Chavakis, Birgit Assmus, Andreas M. Zeiher, and Stefanie Dimmeler
Circulation Research. 2012;111:854-862
Abstract
Rationale: Cell therapy is a promising option for the treatment of acute or chronic myocardial ischemia. The intracoronary infusion of cells imposes the potential risk of cell clotting, which may be prevented by the addition of anticoagulants. However, a comprehensive analysis of the effects of anticoagulants on the function of the cells is missing.
Objective: Here, we investigated the effects of heparin and the thrombin inhibitor bivalirudin on bone marrow–derived mononuclear cell (BMC) functional activity and homing capacity.
Methods and Results: Heparin, but not bivalirudin profoundly and dose-dependently inhibited basal and stromal cell–derived factor 1 (SDF-1)–induced BMC migration. Incubation of BMCs with 20 U/mL heparin for 30 minutes abrogated SDF-1–induced BMC invasion (16±8% of control; P<0.01), whereas no effects on apoptosis or colony formation were observed (80±33% and 100±44% of control, respectively). Pretreatment of BMCs with heparin significantly reduced the homing of the injected cells in a mouse ear-wound model (69±10% of control; P<0.05). In contrast, bivalirudin did not inhibit in vivo homing of BMCs. Mechanistically, heparin binds to both, the chemoattractant SDF-1 and its receptor, chemokine receptor 4 (CXCR4), blocking CXCR4 internalization as well as SDF-1/CXCR4 signaling after SDF-1 stimulation.
Conclusions: Heparin blocks SDF-1/CXCR4 signaling by binding to the ligand as well as the receptor, thereby interfering with migration and homing of BMCs. In contrast, the thrombin inhibitor bivalirudin did not interfere with BMC homing or SDF-1/CXCR4 signaling. These findings suggest that bivalirudin but not heparin might be recommended as an anticoagulant for intracoronary infusion of BMCs for cell therapy after cardiac ischemia.
<img src="http://blogimg.goo.ne.jp/user_image/05/ff/4f10622a72f6316124e2f85e8ec3ce90.png" border="0">
Proposed mechanism. Under normal conditions, stromal cell–derived factor-1 (SDF-1) binds to its receptor, chemokine receptor 4 (CXCR4). The complex internalizes and initiates downstream signaling. Heparin can bind to the chemokine SDF-1, building a heparin–SDF-1 complex. Heparin-bound SDF-1 demonstrates reduced CXCR4 binding ability and thereby impaired downstream signaling. Moreover, heparin can bind directly to the CXCR4 receptor. SDF-1 can still bind to the CXCR4 heparin complex; however, this complex demonstrates reduced internalization and impaired downstream signaling after SDF-1 stimulation.

Reviews
Oral Direct Factor Xa Inhibitors
Calvin H. Yeh, James C. Fredenburgh, and Jeffrey I. Weitz
Circulation Research. 2012;111:1069-1078
Vitamin K antagonists, such as warfarin, have been the mainstay of oral anticoagulation for many decades. Although effective, warfarin has numerous limitations, including a variable dose requirement from patient to patient because of differences in dietary vitamin K intake, common genetic polymorphisms, and multiple drug interactions that affect its pharmacodynamics and metabolism. Consequently, warfarin requires frequent monitoring to ensure that a therapeutic anticoagulant effect has been achieved because excessive anticoagulation can lead to bleeding, and because insufficient anticoagulation can result in thrombosis. Such monitoring is burdensome for patients and physicians and is costly for the health care system. These limitations have prompted the development of new oral anticoagulants that target either factor Xa or thrombin. Although the path to the development of these drugs has been long, the new drugs are at least as effective and safe as warfarin, but they streamline clinical care because they can be administered in fixed doses without routine coagulation monitoring. This article focuses on rivaroxaban, apixaban, and edoxaban, the oral factor Xa inhibitors in the most advanced stages of development. After 20 years of discovery research, these agents are already licensed for several indications. Thus, the long path to finding replacements for warfarin has finally reached fruition. Therefore, development of the oral factor Xa inhibitors represents a translational science success story.

Critical role of factor Xa in coagulation. The extrinsic pathway is initiated when tissue factor (TF) exposed at sites of vascular injury binds activated factor VII (fVIIa) on the activated membrane surface to form extrinsic tenase (Xase). The extrinsic tenase complex activates factors IX and X. Activated factor IX (fIXa) binds to activated factor VIII (fVIIIa) on the activated membrane surface to form the intrinsic tenase complex. IXa also may be generated via the contact pathway after vascular injury. Both the extrinsic and intrinsic tenase complexes generate activated factor X (fXa), which assembles with activated factor V (fVa) on the activated membrane surface to form prothrombinase. The resulting thrombin then converts fibrinogen to fibrin and activates platelets, resulting in the formation of a platelet-fibrin thrombus. By targeting the active site of fXa, the oral fXa inhibitors attenuate thrombin generation that is triggered either via extrinsic tenase or via intrinsic tenase.
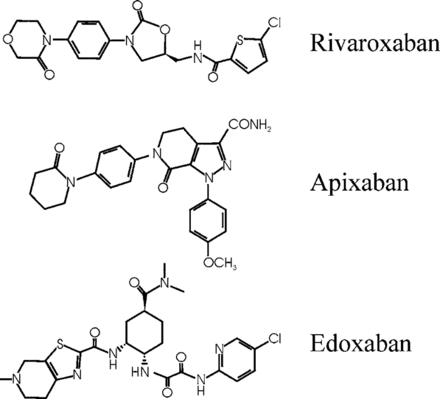
Pharmacological Properties of Warfarin, Rivaroxaban, Apixaban, and Edoxaban
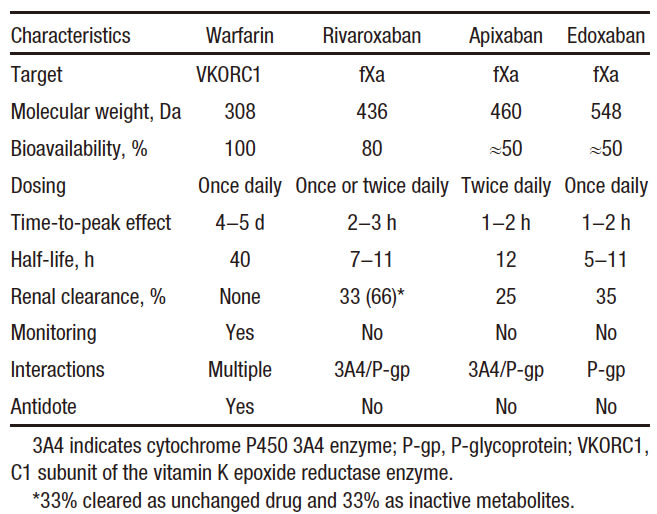
Structures of rivaroxaban, apixaban, and edoxaban, the oral factor Xa inhibitors, in the most advanced stages of development.

Landmark Phase III Clinical Trials and the Number of Patients Enrolled for Rivaroxaban, Apixaban, and Edoxaban for Various Indications
Editorials
G Protein–Coupled Receptor Kinase 5: Exploring Its Hype in Cardiac Hypertrophy
Stephen L. Belmonte and Burns C. Blaxall
Circulation Research. 2012;111:957-958
It is an established dogma that G-protein–coupled receptor kinases (GRKs) classically direct the desensitization and internalization of their eponymous receptors through direct phosphorylation. Yet, it is the noncanonical action of GRKs that has increasingly attracted the interest of groups seeking novel insight into vexing pathophysiological questions. Among the 7 GRK isoforms, several findings suggest that GRK5 may have particular relevance to the development of cardiac hypertrophy and heart failure (HF). Beyond its classical function, GRK5 contains a DNA-binding nuclear localization sequence1 and has recently been reported to modify myocardial gene transcription through histone deacetylase kinase activity.2 Furthermore, GRK5 expression is elevated in the ventricles of patients with HF,3 and transgenic cardiac-specific GRK5 overexpression produced pronounced hypertrophy and accelerated HF progression upon pressure-overload challenge in mice.2
To date, the question remains whether endogenous GRK5 is a prerequisite for hypertrophy and HF development in the face of cardiac stress. In this issue of Circulation Research, Gold et al4 have begun to address this issue by using global and cardiac-restricted GRK5 knockout mice. They report that when subjected to transverse aortic constriction, both the global and cardiac-restricted GRK5 null animals show delayed hypertrophy and preserved heart function compared with control mice, as determined by serial left ventricular posterior wall thickness assessment and ejection fraction, respectively. Furthermore, they used semiquantitative polymerase chain reaction to demonstrate that mRNA expression of a host of hypertrophy marker genes was significantly reduced in the hearts of both GRK5 knockout mice. Similar results were obtained after chronic administration of a subpressor dose of phenylephrine in the global GRK5 knockout mice. Overall, the data suggest that GRK5 is integral to maladaptive cardiac hypertrophy.
In evaluating the results presented in the article by Gold et al,4 it becomes apparent that GRK5 may represent a viable therapeutic target for the treatment of HF, for which new therapies are desperately needed. As with most new and exciting discoveries, there are various facets of GRK5 biology to consider before clinical application. For example, it was previously reported that the global GRK5 knockout mouse exhibits enhanced muscarinic receptor sensitivity but no gross anatomic differences from wild-type littermates.5 In addition, a recent study found high expression of GRK5 in white adipose tissue, underlying reduced adipogenesis and obesity in GRK5-null animals.6 Thus, while cardiac functional parameters clearly suggest a protective role of GRK5 in the heart, it would also be valuable to assess ratios of heart weight to tibia length in addition to body weight, as well as to quantify myocyte size. It is interesting to note that both global and cardiac-restricted GRK5 knockout mice demonstrated progressive, mild (possibly compensatory) cardiac hypertrophy after transverse aortic constriction, whereas the wild-type mice followed the more traditional progression of concentric hypertrophy followed by rapidly decompensated, eccentric hypertrophy coupled to ventricular wall thinning. Attenuation of the hypertrophic gene expression profile after myocardial insult in GRK5 knockouts, partially explained by modest but significant alterations in non-nuclear histone deacetylase phosphorylation, further validates an important role for GRK5 in pathological cardiac hypertrophy.
Importantly, GRK5 is expressed in multiple cardiac cell types. In their current article, Gold et al4 report relatively similar results in both the global and cardiomyocyte-restricted GRK5 mice. Future investigation will be needed to determine the possible functional relevance of GRK5 in the maladaptive hypertrophic response in various nonmyocyte cardiac cells (eg, fibroblasts). Interestingly, prior studies have documented divergent effects of altered GRK5 expression/activity. For example, hybrid transgenic mice overexpressing cardiac GRK5 and a constitutively active α1B-adrenergic receptor mutant demonstrated that GRK5 reduced α1B-adrenergic receptor hypertrophy and partially reduced atrial natriuretic factor mRNA.7 Although somewhat inconsistent with the current report, the inherent differences in the mechanism of injury induced by an activated mutant receptor, transverse aortic constriction, and persistent agonist stimulation may, in large part, explain the discrepancy. It is known that GRK5 phosphorylates and desensitizes α1B-adrenergic receptor basally but not after agonist stimulation.8 Furthermore, the α1B-adrenergic receptor is preferentially expressed in cardiac fibroblasts, whereas the α1A subtype predominates in myocytes.9 It is also likely that GRK5 overexpression confers protection via enhanced desensitization of β-adrenergic receptors, as observed previously in mice.10 This is also the putative explanation for the improved outcomes of patients with HF with a highly active GRK5 polymorphism.11 Considering that GRK5 exhibits differential receptor subtype specificity and has both nuclear and membrane receptor kinase activity, further work is required to establish a definitive role for GRK5 in maladaptive cardiac hypertrophy in a variety of cardiac cell types.
One final point to address is how GRK5 regulates gene transcription. Hypertrophic gene expression is primarily considered in terms of upregulated genes, but downregulated genes are also clinically relevant to hypertrophy and HF. Indeed, it was recently reported that in mice overexpressing Gαq, which produces a pressure-overload cardiac phenotype, enhanced GRK5 expression normalized a subset of downregulated mRNAs responsible for carbohydrate metabolism and energy production.12 Furthermore, expression of a truncated GRK5 that expresses the regulator of G protein signaling homology domain, which inhibits nuclear factor-κB transcriptional activity, reduced left ventricular hypertrophy in spontaneously hypertensive rats or normotensive rats exposed to chronic phenelyephrine.13 Taken together, these results suggest that GRK5 expression counteracts at least some types of hypertrophic stimuli.
In summary, GRK5 expression is clearly pertinent to maladaptive cardiac hypertrophy and the development of HF. Further advances in our understanding of the functional role of GRK5 in heart failure, including those reported by Gold et al,4 may serve to clarify some inconsistencies and take the quest for a new HF therapeutic yet one step closer to reality.
総説 リゾフォスファチジル酸 LPAと炎症
You have free access to this contentLysophosphatidic acid in atherosclerotic diseases (pages 465–482)
Andreas Schober and Wolfgang Siess
Lysophosphatidic acid (LPA) is a potent bioactive phospholipid. As many other biological active lipids, LPA is an autacoid: it is formed locally on demand, and it acts locally near its site of synthesis. LPA has a plethora of biological activities on blood cells (platelets, monocytes) and cells of the vessel wall (endothelial cells, smooth muscle cells, macrophages) that are all key players in atherosclerotic and atherothrombotic processes. The specific cellular actions of LPA are determined by its multifaceted molecular structures, the expression of multiple G-protein coupled LPA receptors at the cell surface and their diverse coupling to intracellular signalling pathways. Numerous studies have now shown that LPA has thrombogenic and atherogenic actions. Here, we aim to provide a comprehensive, yet concise, thoughtful and critical review of this exciting research area and to pinpoint potential pharmacological targets for inhibiting thrombogenic and atherogenic activities of LPA. We hope that the review will serve to accelerate knowledge of basic and clinical science, and to foster drug development in the field of LPA and atherosclerotic/atherothrombotic diseases.
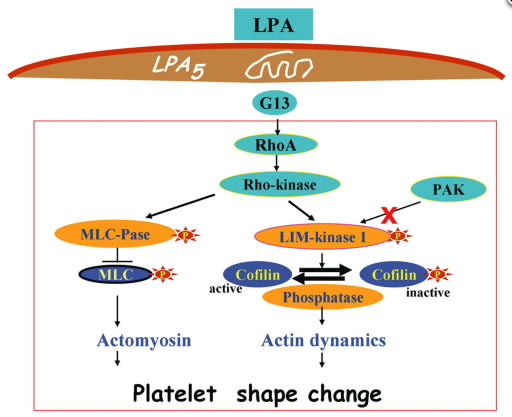
LPA-induced platelet signalling during platelet shape change:理解されている血小板内細胞内情報伝達レベル
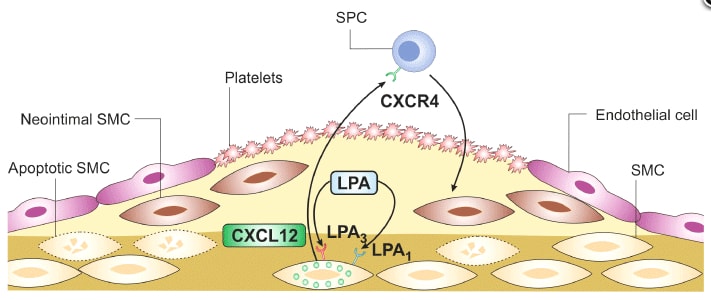
LPA induces neointima formation after vascular injury:理解されている血管新生メカニズム
原著
1.関節炎マウスの大動脈はセロトニンによる収縮反応が悪い「炎症」と「再生」
Contractile, but not endothelial, dysfunction in early inflammatory arthritis: a possible role for matrix metalloproteinase-9 (pages 505–514)
SL Reynolds, AS Williams, H Williams, S Smale, HJ Stephenson, N Amos, SJ George, VB O'Donnell and D Lang
BACKGROUND AND PURPOSE Excess morbidity/mortality in rheumatoid arthritis (RA) is associated with increased incidence of cardiovascular disease. In this ‘proof-of-concept’ study, vascular function was characterized in the murine collagen-induced arthritis (mCIA) model, the benchmark choice for evaluation of the pathological processes and assessment of new therapies.
EXPERIMENTAL APPROACH Mice in the very early stages of arthritis development [and appropriate naïve (non-immunized) age-matched controls] were used in the study. Blood pressure was measured using tail cuff plethysmography. Vascular function in rings of isolated aorta was studied with isometric tension myography. Levels of NO metabolites (NOx), MMP-9 protein and IL-1β in plasma and MMP-9 protein in aortic homogenates were quantified.
KEY RESULTS Impaired vascular contractile responses in arthritis were unaffected by ex vivo inhibition of NOS (endothelial/neuronal and inducible) or COX activities. Endothelium-dependent and -independent relaxation, plasma NOx and blood pressure were unaffected by arthritis. Plasma and aortic homogenate MMP-9 protein levels were increased significantly in arthritis. Incubation of aortic tissues from naïve control animals with exogenous MMP-9 impaired subsequent contractile responses, mirroring that observed in arthritis. A role for IL-1β in perpetuating contractile dysfunction and increasing aortic MMP-9 was excluded.
CONCLUSIONS AND IMPLICATIONS These data identify for the first time a relationship between early arthritis and contractile dysfunction and a possible role for MMP-9 therein, in the absence of overt endothelial dysfunction or increased NO production. As such, MMP-9 may constitute a significant target for early intervention in RA patients with a view to decreasing risk of cardiovascular disease.
炎症期にIL-1β,回復期にMMP9の発現により,セロトニンの収縮反応が低下するという,タイムコース評価の難しい研究系です。
2.Epithelium-dependent modulation of responsiveness of airways from caveolin-1 knockout mice is mediated through cyclooxygenase-2 and 5-lipoxygenase (pages 548–560)
Pawan Sharma, Min H Ryu, Sujata Basu, Sarah A Maltby, Behzad Yeganeh, Mark M Mutawe, Richard W Mitchell and Andrew J Halayko
epartment of Internal Medicine, University of Manitoba, Winnipeg, MB, Canada

BACKGROUND AND PURPOSE Acute silencing of caveolin-1 (Cav-1) modulates receptor-mediated contraction of airway smooth muscle. Moreover, COX-2- and 5-lipoxygenase (5-LO)-derived prostaglandin and leukotriene biosynthesis can influence smooth muscle reactivity. COX-2 half-life can be prolonged through association with Cav-1. We suggested that lack of Cav-1 modulated levels of COX-2 which in turn modulated tracheal contraction, when arachidonic acid signalling was disturbed by inhibition of COX-2.
EXPERIMENTAL APPROACH Using tracheal rings from Cav-1 knockout (KO) and wild-type mice (B6129SF2/J), we measured isometric contractions to methacholine and used PCR, immunoblotting and immunohistology to monitor expression of relevant proteins.
KEY RESULTS Tracheal rings from Cav-1 KO and wild-type mice exhibited similar responses, but the COX-2 inhibitor, indomethacin, increased responses of tracheal rings from Cav-1 KO mice to methacholine. The phospholipase A2 inhibitor, eicosatetraynoic acid, which inhibits formation of both COX-2 and 5-LO metabolites, had no effect on wild-type or Cav-1 KO tissues. Indomethacin-mediated hyperreactivity was ablated by the LTD4 receptor antagonist (montelukast) and 5-LO inhibitor (zileuton). The potentiating effect of indomethacin on Cav-1 KO responses to methacholine was blocked by epithelial denudation. Immunoprecipitation showed that COX-2 binds Cav-1 in wild-type lungs. Immunoblotting and qPCR revealed elevated levels of COX-2 and 5-LO protein, but not COX-1, in Cav-1 KO tracheas, a feature that was prevented by removal of the epithelium.
CONCLUSION AND IMPLICATIONS The indomethacin-induced hypercontractility observed in Cav-1 KO tracheas was linked to increased expression of COX-2 and 5-LO, which probably enhanced arachidonic acid shunting and generation of pro-contractile leukotrienes when COX-2 was inhibited.
3.アデノシンに似ているコルディセピン
Blockade of adipocyte differentiation by cordycepin (pages 561–575)
Shuhei Takahashi, Minori Tamai, Shotaro Nakajima, Hironori Kato, Hisashi Johno, Tomoyuki Nakamura and Masanori Kitamura
BACKGROUND AND PURPOSE Cordyceps militaris has the potential to suppress differentiation of pre-adipocytes. However, the active entities in the extract and the underlying mechanisms of its action are not known. Hence, we investigated whether and how cordycepin (3′-deoxyadenosine), a constituent of C. militaris, inhibits adipogenesis.
EXPERIMENTAL APPROACH Differentiation of 3T3-L1 pre-adipocytes and pre-adipocytes in primary cultures was induced by Insulin, dexamethasone and IBMX, and these were used as in vitro models of adipogenesis. The effects of cordycepin on adipogenesis were examined with particular focus on the regulation of CCAAT/enhancer-binding protein β (C/EBPβ) and PPARγ.
KEY RESULTS Cordycepin suppressed the lipid accumulation and induction of adipogenic markers that occurred on differentiation of pre-adipocytes and also blocked the down-regulation of a pre-adipocyte marker. This anti-adipogenic effect was reversible and mediated by an adenosine transporter, but not A1, A2 or A3 adenosine receptors. This effect of cordycepin was not reproduced by other adenosine-related substances, including ATP, ADP and adenosine. Early induction of the adipogenic C/EBPβ–PPARγ pathway was suppressed by cordycepin. Blockade of mTORC1 via inhibition of PKB (Akt) and activation of AMP kinase was identified as the crucial upstream event targeted by cordycepin. In addition to its negative effect on adipogenesis, cordycepin suppressed lipid accumulation in mature adipocytes.
CONCLUSIONS AND IMPLICATIONS These results suggest that the anti-adipogenic effects of cordycepin occur through its intervention in the mTORC1-C/EBPβ–PPARγ pathway. Cordycepin, by blocking both adipogenesis and lipid accumulation, may have potential as a therapeutic agent for effective treatment of obesity and obesity-related disorders.
mTOR複合体1への作用が重要
1)コルディセピンは,「さなぎ」に含まれています。「さなぎ」の粉末は,腸粘膜新生を抑制,がん細胞増殖を抑制などの作用があるとされています。
2)PKB活性を阻害する可能性があり,mTORC1を抑制する作用があります。
3)コルディセピンは,C/EBP mRNAを低下させる可能性があります。
4.Toll-like receptor 4 knockout protects against anthrax lethal toxin-induced cardiac contractile dysfunction: role of autophagy (pages 612–626)
Machender R Kandadi, Arthur E Frankel and Jun Ren 競合する研究内容
BACKGROUND AND PURPOSE Anthrax lethal toxin (LeTx) is known to induce circulatory shock and death, although the underlying mechanisms have not been elucidated. This study was designed to evaluate the role of toll-like receptor 4 (TLR4) in anthrax lethal toxin-induced cardiac contractile dysfunction.
EXPERIMENTAL APPROACH Wild-type (WT) and TLR4 knockout (TLR-/-) mice were challenged with lethal toxin (2 µg·g-1, i.p.), and cardiac function was assessed 18 h later using echocardiography and edge detection. Small interfering RNA (siRNA) was employed to knockdown TLR4 receptor or class III PI3K in H9C2 myoblasts. GFP–LC3 puncta was used to assess autophagosome formation. Western blot analysis was performed to evaluate autophagy (LC3, Becline-1, Agt5 and Agt7) and endoplasmic reticulum (ER) stress (BiP, eIF2α and calreticulin).
KEY RESULTS In WT mice, lethal toxin exposure induced cardiac contractile dysfunction, as evidenced by reduced fractional shortening, peak shortening, maximal velocity of shortening/re-lengthening, prolonged re-lengthening duration and intracellular Ca2+ derangement. These effects were significantly attenuated or absent in the TLR4 knockout mice. In addition, lethal toxin elicited autophagy in the absence of change in ER stress. Knockdown of TLR4 or class III PI3 kinase using siRNA but not the autophagy inhibitor 3-methyladenine significantly attenuated or inhibited lethal toxin-induced autophagy in H9C2 cells.
CONCLUSION AND IMPLICATIONS Our results suggest that TLR4 may be pivotal in mediating the lethal cardiac toxicity induced by anthrax possibly through induction of autophagy. These findings suggest that compounds that negatively modulate TLR4 signalling and autophagy could be used to treat anthrax infection-induced cardiovascular complications.
1. PM10による肺炎症に対するロバスタチンの抑制効果;Urban air PM10 (EHC-93) from Environmental Health Directorate, Health Canada (Ottawa, ON, Canada)
Ryohei Miyata, Ni Bai, Renaud Vincent, Don D. Sin, and Stephan F. Van Eeden
Novel properties of statins: suppression of the systemic and bone marrow responses induced by exposure to ambient particulate matter (PM10) air pollution
Am J Physiol Lung Cell Mol Physiol September 15, 2012 303:L492-L499

Stephanus (Stephan) F. van Eeden MD, PhD, FRCPC(C)
Department of Internal Medicine, University of British Columbia
Abstract
Exposure to ambient particulate matter (PM10) elicits systemic inflammatory responses that include the stimulation of bone marrow and progression of atherosclerosis. The present study was designed to assess the effect of repeated exposure of PM10 on the turnover and release of polymorphonuclear leukocytes (PMNs) from the bone marrow into the circulation and the effect of lovastatin on the PM10-induced bone marrow stimulation. Rabbits exposed to PM10 three times a week for 3 wk, were given a bolus of 5′-bromo-2′-deoxyuridine to label dividing cells in the marrow to calculate the transit time of PMNs in the mitotic or postmitotic pool. PM10 exposure accelerated the turnover of PMNs by shortening their transit time through the marrow (64.8 ± 1.9 h vs. 34.3 ± 7.4 h, P < 0.001, control vs. PM10). This was predominantly due to a rapid transit of PMNs through the postmitotic pool (47.9 ± 0.7 h vs. 21.3 ± 4.3 h, P < 0.001, control vs. PM10) but not through the mitotic pool. Lovastatin delayed the transit time of postmitotic PMNs (38.2 ± 0.5 h, P < 0.001 vs. PM10) and shifted the postmitotic PMN release peak from 30 h to 48 h. PM10 exposure induced the prolonged retention of newly released PMNs in the lung, which was reduced by lovastatin (P < 0.01). PM10 exposure increased plasma interleukin-6 levels with significant reduction by lovastatin (P < 0.01). We conclude that lovastatin downregulates the PM10-induced overactive bone marrow by attenuating PM10.
MANY WELL-DOCUMENTED STUDIES have supported the concept that ambient particulate matter (particles less than 10 μm or PM10) exposure induces systemic inflammatory responses, characterized by an increase in circulating proinflammatory mediators and blood elements such as leukocytes or platelets. These systemic responses to PM10 exposure have been implicated in the risk for downstream cardiovascular events. The focus of their research efforts are the mechanisms of lung inflammation, particularly, lung inflammation caused by infection, cigarette smoking and air pollution.
2. 腸管由来のセロトニンが肺高血圧を緩和している可能性
Shariq Abid, Amal Houssaini, Caroline Chevarin, Elisabeth Marcos, Claire-Marie Tissot, Guillaume Gary-Bobo, Feng Wan, Nathalie Mouraret, Valerie Amsellem, Jean-Luc Dubois-Randé, Michel Hamon, and Serge Adnot
Inhibition of gut- and lung-derived serotonin attenuates pulmonary hypertension in mice
Am J Physiol Lung Cell Mol Physiol September 15, 2012 303:L500-L508

Serge Adnot, MD, PhD
Medical School of Creteil
Hôpital Henri Mondor
Créteil, France
Dr. Adnot is a Professor in the Department of Physiology at the Medical school of Créteil, and Cardiologist and Head of Medical Department of Functional Testing at Hôpital Henri Mondor, and Director of INSERM Research Unit 651 (2005).
Abstract
Decreasing the bioavailability of serotonin (5-HT) by inhibiting its biosynthesis may represent a useful adjunctive treatment of pulmonary hypertension (PH). We assessed this hypothesis using LP533401, which inhibits the rate-limiting enzyme tryptophan hydroxylase 1 (Tph1) expressed in the gut and lung, without inhibiting Tph2 expressed in neurons. Mice treated repeatedly with LP533401 (30–250 mg/kg per day) exhibited marked 5-HT content reductions in the gut, lungs, and blood, but not in the brain. After a single LP533401 dose (250 mg/kg), lung and gut 5-HT contents decreased by 50%, whereas blood 5-HT levels remained unchanged, suggesting gut and lung 5-HT synthesis. Treatment with the 5-HT transporter (5-HTT) inhibitor citalopram decreased 5-HT contents in the blood and lungs but not in the gut. In transgenic SM22-5-HTT+ mice, which overexpress 5-HTT in smooth muscle cells and spontaneously develop PH, 250 mg/kg per day LP533401 or 10 mg/kg per day citalopram for 21 days markedly reduced lung and blood 5-HT levels, right ventricular (RV) systolic pressure, RV hypertrophy, distal pulmonary artery muscularization, and vascular Ki67-positive cells (P < 0.001). Combined treatment with both drugs was more effective in improving PH-related hemodynamic parameters than either drug alone. LP533401 or citalopram treatment partially prevented PH development in wild-type mice exposed to chronic hypoxia. Lung and blood 5-HT levels were lower in hypoxic than in normoxic mice and decreased further after LP533401 or citalopram treatment. These results provide proof of concept that inhibiting Tph1 may represent a new therapeutic strategy for human PH.
Tryptophan Hydroxylase は,セロトニン合成酵素であり,(トリプトファンの5位にヒドロキシル基をふかし,5-ヒドロキシトリプトファン(5-HTP)を形成する。この5-HTPがセロトニン合成の重要な律速酵素となる。Tph1とThp2つのサブタイプが,別々な遺伝子デコードされている。このうち,Thp-1は消化管や肺や精巣に,Thp-2はニューロンに比較的高い特異性を持って存在しているようだ。この研究で用いられているLP533401が,比較的選択性の高いTph1阻害薬である。
3. Alveolar Fluid Clearance (AFC)
Yosaf F. Zeyed, Julie A. Bastarache, Michael A. Matthay, and Lorraine B. Ware
The severity of shock is associated with impaired rates of net alveolar fluid clearance in clinical acute lung injury
Am J Physiol Lung Cell Mol Physiol September 15, 2012 303:L550-L555

Abstract
The rate of alveolar fluid clearance (AFC) is associated with mortality in clinical acute lung injury (ALI). Patients with ALI often develop circulatory shock, but how shock affects the rate of AFC is unknown. To determine the effect of circulatory shock on the rate of AFC in patients with ALI, the rate of net AFC was measured in 116 patients with ALI by serial sampling of pulmonary edema fluid. The primary outcome was the rate of AFC in patients with shock compared with those without shock. We also tested the effects of shock severity and bacteremia. Patients with ALI and shock (n = 86) had significantly slower rates of net AFC compared with those without shock (n = 30, P = 0.03), and AFC decreased significantly as the number of vasopressors increased. Patients with positive blood cultures (n = 21) had slower AFC compared with patients with negative blood cultures (n = 96, P = 0.023). In addition, the edema fluid-to-plasma protein ratio, an index of alveolar-capillary barrier permeability, was highest in patients requiring the most vasopressors (P < 0.05). Patients with ALI complicated by circulatory shock and bacteremia had slower rates of AFC compared with patients without shock or bacteremia. An impaired capacity to reabsorb alveolar edema fluid may contribute to high mortality among patients with sepsis-induced ALI. These findings also suggest that vasopressor use may be a marker of alveolar-capillary barrier permeability in ALI and provide justification for new therapies that enhance alveolar epithelial and endothelial barrier integrity in ALI, particularly in patients with shock.
※ Lorraine B. Wareのエディトリアル
Modeling human lung disease in animals
Am J Physiol Lung Cell Mol Physiol February 2008 294:L149-L150
Overview of Pyridine Nucleotides Review Series
Michinari Nakamura, Aruni Bhatnagar, and Junichi Sadoshima
Circulation Research. 2012;111:604-610
Regulation of Cell Survival and Death by Pyridine Nucleotides
Shin-ichi Oka, Chiao-Po Hsu, and Junichi Sadoshima
Circulation Research. 2012;111:611-627
Pyridine Nucleotide Regulation of Cardiac Intermediary Metabolism
John R. Ussher, Jagdip S. Jaswal, and Gary D. Lopaschuk
Circulation Research. 2012;111:628-641
Sirtuins and Pyridine Nucleotides
Maha Abdellatif
Circulation Research. 2012;111:642-656
総説 血管内皮細胞のTRPA1について
You have free access to this contentTRPA1 channels in the vasculature (pages 13–22)
Scott Earley
Article first published online: 3 AUG 2012 | DOI: 10.1111
Abstruct
This review is focused on the role of the ankyrin (A) transient receptor potential (TRP) channel TRPA1 in vascular regulation. TRPA1 is activated by environmental irritants, pungent compounds found in foods such as garlic, mustard and cinnamon, as well as metabolites produced during oxidative stress. The structure of the channel is distinguished by the ∼14–19 ankyrin repeat (AR) domains present in the intracellular amino terminus. TRPA1 has a large unitary conductance (98 pS) and slight selectivity for Ca2+ versus Na+ ions (PCa/PNa ≈ 7.9). TRPA1 is involved in numerous important physiological processes, including nociception, mechanotransduction, and thermal and oxygen sensing. TRPA1 agonists cause arterial dilation through two distinctive pathways. TRPA1 channels present in perivascular nerves mediate vasodilatation of peripheral arteries in response to chemical agonists through a mechanism requiring release of calcitonin gene-related peptide. In the cerebral circulation, TRPA1 channels are present in the endothelium, concentrated within myoendothelial junction sites. Activation of TRPA1 channels in this vascular bed causes endothelium-dependent smooth muscle cell hyperpolarization and vasodilatation that requires the activity of small and intermediate conductance Ca2+-activated K+ channels. Systemic administration of TRPA1 agonists causes transient depressor responses, followed by sustained increases in heart rate and blood pressure that may result from elevated sympathetic nervous activity. These findings indicate that TRPA1 activity influences vascular function, but the precise role and significance of the channel in the cardiovascular system remains to be determined.
Transient Receptor Potential superfamily について
The mammalian transient receptor potential (TRP) superfamily of cation channels comprises 28 members assigned to six subfamilies based on sequence homology. The ankyrin (A) subfamily is the smallest and is composed of only a single member, TRPA1 (originally designated as ANKTM1). Despite being one of the last TRP channels to be discovered (Story et al., 2003), TRPA1 has garnered a great deal of recent attention. The essential properties and structure of TRPA1 have been evolutionarily conserved for more than 500 million years (Kang et al., 2010). The channel likely evolved as a sensor of electrophilic toxicity (Kang et al., 2010) before divergent specialized functions developed in different species (Story et al., 2003; Rosenzweig et al., 2005; Cordero-Morales et al., 2011; Geng et al., 2011). TRPA1, like all TRP channels, is expressed as six-transmembrane domain polypeptide subunits, a motif common to many types of ion channels. Functional TRPA1 channels are formed from four of these subunits. Assembled TRPA1 channels are thought to have a homomeric structure (composed of four identical subunits), as there is currently no evidence that heteromultimeric channels involving other TRP channel subunits can form. The channel is distinguished structurally by, and named for, the ∼14–19 ankyrin repeat (AR) domains forming a portion of the protein's intracellular N-terminus. In general, AR domains mediate protein–protein interactions and provide mechanical elasticity (Sedgwick and Smerdon, 1999), although a recent study suggests that particular TRPA1 AR domains can regulate agonist- and heat-induced channel activity (Cordero-Morales et al., 2011). TRPA1 was originally described as a non-selective cation channel that is equally permeable to Na+ versus Ca2+ ions (PCa/PNa reported as 0.84–3.28) (Story et al., 2003; Wang et al., 2008), although Karashima et al. found that during agonist stimulation, PCa/PNa = 7.91 ± 0.60 and the fractional Ca2+ current under these conditions is 17.9–22.3% (Karashima et al., 2010). The unitary conductance of the channel is large (98 pS, when physiological ionic gradients are maintained) (Nagata et al., 2005), indicating that TRPA1 channels can support consequential levels of Ca2+ influx. Predictably, TRPA1 has been shown to influence a broad range of physiological processes that involve Ca2+-dependent signalling pathways, including nociception, mechanotransduction, thermal and oxygen sensing, and responses to environmental irritants and pungent compounds. This manuscript focuses on the role of TRPA1 channels in vascular regulation. The relevant pharmacology is discussed, and studies investigating the consequences of TRPA1 activity on local and integrative control of the vasculature are reviewed.
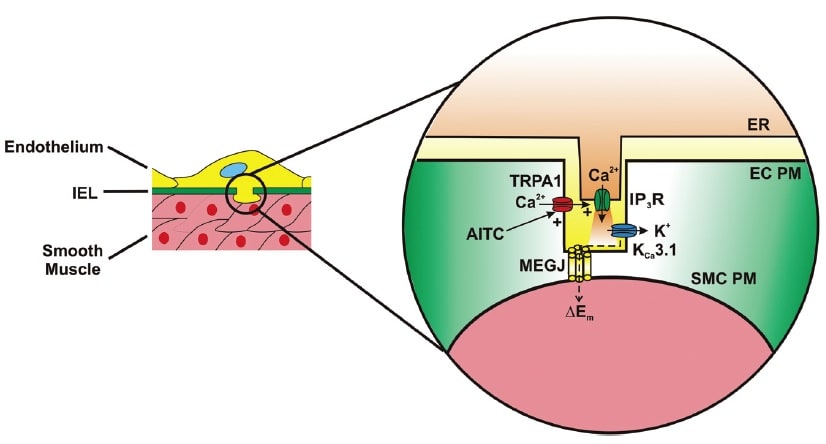
Activation of TRPA1 in cerebral arteries causes endothelium-dependent vasodilation. Allyl isothiocyanate (AITC) activates Ca2+ influx via TRPA1 channels present in myoendothelial junctions in cerebral arteries. TRPA1-mediated Ca2+ influx stimulates Ca2+ release from the endoplasmic reticulum (ER) via inositol trisphosphate receptors (IP3R). The resulting Ca2+ signal (i.e. Ca2+ pulsar) stimulates proximal intermediate conductance Ca2+-activated K+ channels (KCa3.1), resulting in hyperpolarization of the endothelial cell plasma membrane (EC PM). The change in membrane potential (DEm) is conducted via myoendothelial gap junctions (MEGJs) to hyperpolarize the vascular smooth muscle cell plasma membrane (SMCPM), resulting in myocyte relaxation.

Activation of TRPA1 channels in sensory nerves causes arterial dilation. Allyl isothiocyanate (AITC), allicin, cinnamaldehyde (CA) and 4-oxo-2-nonenal (4-ONE) activate Ca2+ influx via TRPA1 channels in sensory nerves, causing release of calcitonin gene-related peptide (CGRP) from perivascular terminals. CGRP binds to its G protein-coupled receptor (GPCR) on the plasma membrane of vascular smooth muscle cells (SMCs) to cause membrane hyperpolarization and myocyte relaxation.
RESEARCH PAPERS
1. メラノコルチン受容体MC1とMC3を介した炎症の保護作用 <炎症のタイムコースが大切>
Chondroprotective and anti-inflammatory role of melanocortin peptides in TNF-α activated human C-20/A4 chondrocytes (pages 67–79)
Magdalena K Kaneva, Mark JP Kerrigan, Paolo Grieco, G Paul Curley, Ian C Locke and Stephen J Getting
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.
BACKGROUND AND PURPOSE
Melanocortin MC1 and MC3 receptors, mediate the anti-inflammatory effects of melanocortin peptides. Targeting these receptors could therefore lead to development of novel anti-inflammatory therapeutic agents. We investigated the expression of MC1 and MC3 receptors on chondrocytes and the role of α-melanocyte-stimulating hormone (α-MSH) and the selective MC3 receptor agonist, [DTRP8]-γ-MSH, in modulating production of inflammatory cytokines, tissue-destructive proteins and induction of apoptotic pathway(s) in the human chondrocytic C-20/A4 cells.
EXPERIMENTAL APPROACH
Effects of α-MSH, [DTRP8]-γ-MSH alone or in the presence of the MC3/4 receptor antagonist, SHU9119, on TNF-α induced release of pro-inflammatory cytokines, MMPs, apoptotic pathway(s) and cell death in C-20/A4 chondrocytes were investigated, along with their effect on the release of the anti-inflammatory cytokine IL-10.
KEY RESULTS
C-20/A4 chondrocytes expressed functionally active MC1,3 receptors. α-MSH and [DTRP8]-γ-MSH treatment, for 30 min before TNF-α stimulation, provided a time-and-bell-shaped concentration-dependent decrease in pro-inflammatory cytokines (IL-1β, IL-6 and IL-8) release and increased release of the chondroprotective and anti-inflammatory cytokine, IL-10, whilst decreasing expression of MMP1, MMP3, MMP13 genes.α-MSH and [DTRP8]-γ-MSH treatment also inhibited TNF-α-induced caspase-3/7 activation and chondrocyte death. The effects of [DTRP8]-γ-MSH, but not α-MSH, were abolished by the MC3/4 receptor antagonist, SHU9119.
CONCLUSION AND IMPLICATIONS
Activation of MC1/MC3 receptors in C-20/A4 chondrocytes down-regulated production of pro-inflammatory cytokines and cartilage-destroying proteinases, inhibited initiation of apoptotic pathways and promoted release of chondroprotective and anti-inflammatory cytokines. Developing small molecule agonists to MC1/MC3 receptors could be a viable approach for developing chondroprotective and anti-inflammatory therapies in rheumatoid and osteoarthritis.
2. PAR1 inhibitor Q94/Q109について
Modulation of PAR1 signalling by benzimidazole compounds (pages 80–94)
S Asteriti, S Daniele, F Porchia, MT Dell' Anno, A Fazzini, I Pugliesi, ML Trincavelli, S Taliani, C Martini, MR Mazzoni and A Gilchrist
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.
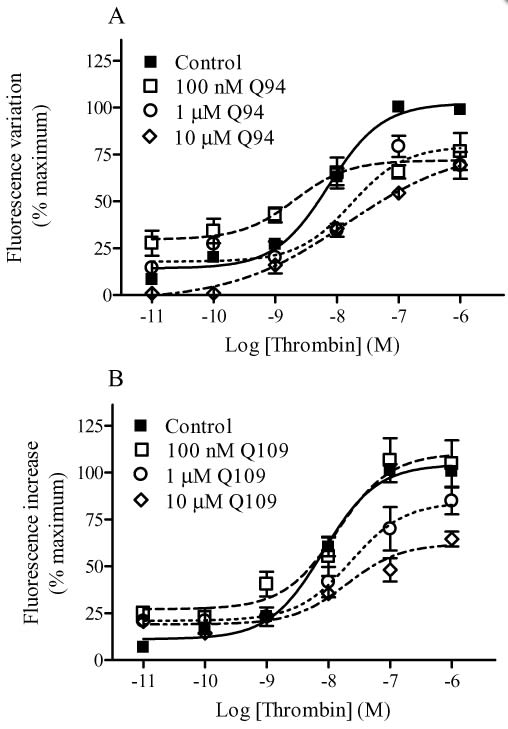
Modulation of thrombin-induced intracellular Ca2+ mobilization by Q94 (A) and Q109 (B). Intracellular Ca2+ mobilization was measured using Fluo 3-AM-loaded HMECs-1 as described in Methods. Basal [Ca2+]i was 240.1 ± 45.3 nM (n = 4). The benzimidazole compounds were added 15 min before the addition of thrombin. The concentration–response curves were performed using seven different concentrations of the enzyme in the presence and absence of fixed concentrations of PAR1 modulators. Data are reported as % of maximal RF and are the means ± SEM of three (A) and four (B) independent experiments, each performed in triplicate.
研究内容:
BACKGROUND AND PURPOSE
Recently, a small molecule (Q94) was reported to selectively block PAR1/Gαq interaction and signalling. Here, we describe the pharmacological properties of Q94 and two analogues that share its benzimidazole scaffold (Q109, Q89). Q109 presents a modest variation from Q94 in the substituent group at the 2-position, while Q89 has quite different groups at the 1- and 2-positions.
EXPERIMENTAL APPROACH
Using human microvascular endothelial cells, we examined intracellular Ca2+ mobilization and inositol 1,4,5-trisphosphate accumulation as well as isoprenaline- or forskolin-stimulated cAMP production in response to thrombin.
KEY RESULTS
Q89 (10 µM) produced a leftward shift in the thrombin-mediated intracellular Ca2+ mobilization concentration–response curve while having no effect on the Emax. Both Q94 (10 µM) and Q109 (10 µM) reduced intracellular Ca2+ mobilization, leading to a decrease in Emax and an increase in EC50 values. Experiments utilizing receptor-specific activating peptides confirmed that Q94 and Q109 were selective for PAR1 as they did not alter the Ca2+ response mediated by a PAR2 activating peptide. Consistent with our Ca2+ results, micromolar concentrations of either Q94 or Q109 significantly reduced thrombin-induced inositol 1,4,5-trisphosphate production. Neither Q94 nor Q109 diminished the inhibitory effects of thrombin on cAMP production, indicating they inhibit signalling selectively through the Gq pathway. Our results also suggest the 1,2-disubstituted benzimidazole derivatives act as ‘allosteric agonists’ of PAR1.
CONCLUSIONS AND IMPLICATIONS
The Q94 and Q109 benzimidazole derivatives represent a novel scaffold for the development of new PAR1 inhibitors and provide a starting point to develop dual signalling pathway-selective positive/negative modulators of PAR1.
Introductionの記載
Protease-activated receptors (PARs) are a family of four GPCRs (PAR1, PAR2, PAR3, and PAR4; receptor nomenclature follows Alexander et al., 2011) characterized by a unique mechanism of activation. PARs are activated enzymatically through proteolysis of the receptor by enzymes of the serine protease family (Macfarlane et al., 2001). The proteolytic cleavage occurs at specific sites within their N-terminal region, thereby exposing novel N-termini, and the ‘tethered ligand’ then folds back onto the extracellular loop II of the receptor, resulting in activation. PAR1, PAR3, and PAR4 are preferentially cleaved by thrombin; whereas PAR2 is mainly a substrate for trypsin, and mast cell tryptase (Coughlin, 2001; Macfarlane et al., 2001; Hollenberg and Compton, 2002). In addition to proteolytic cleavage, most PARs can be activated by synthetic peptides corresponding to the tethered ligand (TL) sequence (Ramachandran and Hollenberg, 2008).
PARs are expressed in many cell types and different organ systems. For example, PAR1, PAR2, PAR3 and PAR4 are all expressed on human endothelial cells (Ramachandran and Hollenberg, 2008); although PAR4 expression may be localized to the endothelium of specific vascular areas (O'Brien et al., 2000; Fujiwara et al., 2005; Hirano et al., 2007). Upon activation, PAR1 exerts its effects on endothelium by activating multiple G-proteins, including Gi/o, Gq/11 and G12/13, leading to modulation of numerous downstream signalling pathways (Barr et al., 1997; Vanhauwe et al., 2002; Ramachandran and Hollenberg, 2008). While PAR1 expression is widely distributed among cells and tissues, PAR2 expression is more limited, and studies indicate that signalling occurs via Gq/11, Gi/o (Nystedt et al., 1995; Macfarlane et al., 2001) and perhaps G12/13 (Ramachandran et al., 2009).
PAR activation plays a key role in many physiological and pathophysiological events involving different organ systems (Ramachandran and Hollenberg, 2008). For example, in the cardiovascular and circulatory systems, activation of PAR1 and to a lesser extent PAR4 on human platelets is sufficient to trigger aggregation (Kahn et al., 1999), while activation of human endothelial PAR1 and PAR2 causes vascular relaxation (Hamilton et al., 2001; 2002; Robin et al., 2003). Indeed, PAR antagonists might prove useful therapeutically for the treatment of several diseases, including thrombosis and atherosclerosis.
Several peptide, peptidomimetic and non-peptide PAR1 antagonists are currently available for experimental studies; and a number of synthetic small molecules are being evaluated for pharmaceutical use in humans (reviewed by Chackalamannil, 2006). In addition, alternative approaches to inhibit PAR1 signalling, such as transfection of endothelial cells with minigene vectors expressing Gα carboxyl (C)-terminal peptides (Gilchrist et al., 2001), or the use of membrane-permeable peptides termed ‘pepducins’ derived from the sequence of the third intracellular loop of PAR1 (Covic et al., 2002a,b), have been presented. Deng et al. (2008) reported the use of a small molecule, Q94, which selectively blocks the interaction between PAR1 and Gαq, to investigate thrombin mediated signalling in mouse lung fibroblasts. Q94 was originally identified during an elisa screen for competition of a high-affinity peptide mimicking the C-terminus of Gαq using a compound library (Deng et al., 2008), and the compound may act as a negative allosteric modulator of PAR1 rather than an orthosteric antagonist. Although Q94 has not been extensively investigated, it represents the first compound to show selective modulation of PAR1/G-protein interactions and thus serve as a biased inhibitor of thrombin-mediated Gq pathway signalling events.
In the present study, we examined the pharmacological properties of Q94 and two analogues, Q109 and Q89, using human microvascular endothelial cells. The three small molecules (Q94, Q109, Q89) all share a benzimidazole scaffold and present either a modest variation in the substituent group at the 2-position (Q109 vs. Q94) or quite different groups at the 1- and 2-positions (Q89 vs. Q94). Whereas micromolar concentrations of Q94 or Q109 resulted in a 30–50% reduction of thrombin's maximal effect (Emax) on intracellular Ca2+ mobilization in combination with a two- to threefold increase of thrombin's EC50 value, a micromolar concentration of Q89 produced a shift to the left in thrombin's concentration–response curve and had no effect on thrombin Emax. Similar to the Ca2+ mobilization studies, experiments assessing inositol-1,4,5-trisphosphate (IP3) accumulation indicated that the presence of micromolar concentrations of either Q94 or Q109 resulted in a significant decrease in thrombin's maximal stimulation (Emax). The antagonistic properties of Q94 and Q109 appear selective as they affected the concentration–response curve of a selective PAR1 activating peptide (AP) while not altering that of a selective PAR2-AP. In addition, these benzimidazole derivatives did not reverse the inhibitory effect of thrombin on isoprenaline- or forskolin-stimulated cAMP production, suggesting they can selectively inhibit the Gq pathway. Importantly, our studies reveal that although the benzimidazole derivatives Q94 and Q109 behave as selective modulators of PAR1 signalling and represent a novel scaffold for the development of new PAR1 inhibitors, their effect on PAR1 signalling is more complex than simple inhibition of Gq activation.
3. IKKβの阻害について
1-Dehydro-[10]-gingerdione from ginger inhibits IKKβ activity for NF-κB activation and suppresses NF-κB-regulated expression of inflammatory genes (pages 128–140)
Hwa Young Lee, Sun Hong Park, Misoon Lee, Hye-Jin Kim, Shi Yong Ryu, Nam Doo Kim, Bang Yeon Hwang, Jin Tae Hong, Sang-Bae Han and Youngsoo Kim
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.
注意事項
Genetic substitutions of the activation loop Ser177 and Ser181 residues with Ala decrease IKKβ activity, whereas those of IKKβ (SS/EE) stimulate kinase activity by mimicking Ser phosphorylation (Mercurio et al., 1997; 1999). Interestingly, IKKβ (C/A) decreases its kinase activity, suggesting that Cys179 plays an important role in the binding affinity of cofactor ATP with IKKβ and in the Ser phosphorylation of IKKβ to stimulate its kinase activity (Byun et al., 2006). To further understand the molecular mechanism of D10G, we proposed molecular docking of D10G to the crystal structure of human IKKβ. The activation loop of IKKβ was somewhat flexible by itself but subtly rearranged to an extended structure upon irreversible binding of D10G with the Cys179 under the most energetically favourable simulation. This conformational change might contribute to the inhibitory mechanism of D10G on IKKβ activity.
BACKGROUND AND PURPOSE
Pungent constituents of ginger (Zingiber officinale) have beneficial effects on inflammatory pain and arthritic swelling. However, the molecular basis for these pharmacological properties is only partially understood. Here, we investigated the molecular target of 1-dehydro-[10]-gingerdione (D10G), one of the pungent constituents of ginger, that mediates its suppression of NF-κB-regulated expression of inflammatory genes linked to toll-like receptor (TLR)-mediated innate immunity.
EXPERIMENTAL APPROACH
RAW 264.7 macrophages or primary macrophages-derived from bone marrows of C57BL/6 or C3H/HeJ mice were stimulated with the TLR4 agonist LPS in the presence of D10G. Catalytic activity of inhibitory κB (IκB) kinase β (IKKβ) was determined by a kinase assay and immunoblot analysis, and the expression of inflammatory genes by RT-PCR analysis and a promoter-dependent reporter assay.
KEY RESULTS
D10G directly inhibited the catalytic activity of cell-free IKKβ. Moreover, D10G irreversibly inhibited cytoplasmic IKKβ-catalysed IκBα phosphorylation in macrophages activated by TLR agonists or TNF-α, and also IKKβ vector-elicited NF-κB transcriptional activity in these cells. These effects of D10G were abolished by substitution of the Cys179 with Ala in the activation loop of IKKβ, indicating a direct interacting site of D10G. This mechanism was shown to mediate D10G-induced disruption of NF-κB activation in LPS-stimulated macrophages and the suppression of NF-κB-regulated gene expression of inducible NOS, COX-2 and IL-6.
CONCLUSION AND IMPLICATIONS
This study demonstrates that IKKβ is a molecular target of D10G involved in the suppression of NF-κB-regulated gene expression in LPS-activated macrophages; this suggests D10G has therapeutic potential in NF-κB-associated inflammation and autoimmune disorders.
彼らのMethods記載
Cell culture
RAW 264.7 macrophages were purchased from ATCC (Manassas, VA). Primary macrophages were prepared from bone marrows of C57BL/6 or C3H/HeJ mice as described previously (Chung et al., 2010). All studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010;McGrath et al., 2010). Animal experiments were carried out following the protocols approved by Animal Experimentation Ethics Committee in CBNU institute. Macrophages were grown in DMEM containing 10% FBS, benzylpenicillin potassium (143 U·mL-1) and streptomycin sulfate (100 µg·mL-1) at 37°C and 5% CO2. RAW 267.4 cells harbuoring the pNF-κB-SEAP-NPT construct were cultured in the same media with an additional supplement of geneticin (500 µg·mL-1).
IKKβ kinase assay
Ser/Thr kinase activity of IKKβ was determined as described previously (Kim et al., 2008). In brief, IKKβ proteins were reacted with substrate GST-IκB (2 µg) and co-factor [γ-32P]-ATP (5 µCi) in a kinase buffer (20 mM HEPES, pH 7.7, 2 mM MgCl2, 50 µM ATP, 10 mM β-glycerophosphate, 10 mM NaF, 300 µg·mL-1 Na3VO4, 2 µM PMSF, 10 µg·mL-1 aprotinin, 1 µg·mL-1 leupeptin, 1 µg·mL-1 pepstatin) at 30°C for 1 h. These reaction mixtures were resolved on SDS-acrylamide gels by electrophoresis. Radioactive bands from the dried gels were then visualized by exposure to X-ray film. Wild-type IKKβ proteins were purchased from Millipore (Billerica, MA, USA). For preparing point-substituted IKKβ proteins, RAW 264.7 cells were transfected with FLAG-tagged expression vector encoding IKKβ (SS/EE) with Glu residues instead of Ser177 and Ser181 or IKKβ (C/A) with Ala residue instead of Cys179 (Kim et al., 2008). Point-substituted IKKβ proteins were precipitated from cell extracts using anti-FLAG affinity gel freezer-safe beads, washed with 20 mM HEPES (pH 7.7) and then subjected to the kinase assay.
4. 鮭カルシトニンでDM治療
Oral salmon calcitonin attenuates hyperglycaemia and preserves pancreatic beta-cell area and function in Zucker diabetic fatty rats (pages 151–163)
M Feigh, KV Andreassen, AV Neutzsky-Wulff, ST Petersen, C Hansen, AC Bay-Jensen, JE Henriksen, H Beck-Nielsen, C Christiansen, K Henriksen and MA Karsdal
Article first published online: 3 AUG 2012 | DOI: 10.1111/j.1476-5381.2012.
BACKGROUND AND PURPOSE
Oral salmon calcitonin (sCT), a dual-action amylin and calcitonin receptor agonist, improved glucose homeostasis in diet-induced obese rats. Here, we have evaluated the anti-diabetic efficacy of oral sCT using parameters of glycaemic control and beta-cell morphology in male Zucker diabetic fatty (ZDF) rats, a model of type 2 diabetes.
EXPERIMENTAL APPROACH
Male ZDF rats were treated with oral sCT (0.5, 1.0 or 2 mg·kg-1) or oral vehicle twice daily from age 8 to 18 weeks. Zucker lean rats served as control group. Fasting and non-fasted blood glucose, glycosylated haemoglobin (HbA1c) and levels of pancreas and incretin hormones were determined. Oral glucose tolerance test and i.p. glucose tolerance test were compared, and beta-cell area and function were evaluated.
KEY RESULTS
Oral sCT treatment dose-dependently attenuated fasting and non-fasted hyperglycaemia during the intervention period. At the end of the study period, oral sCT treatment by dose decreased diabetic hyperglycaemia by ∼9 mM and reduced HbA1c levels by 1.7%. Furthermore, a pronounced reduction in glucose excursions was dose-dependently observed for oral sCT treatment during oral glucose tolerance test. In addition, oral sCT treatment sustained hyperinsulinaemia and attenuated hyperglucagonaemia and hypersecretion of total glucagon-like peptide-1 predominantly in the basal state. Lastly, oral sCT treatment dose-dependently improved pancreatic beta-cell function and beta-cell area at study end.
CONCLUSIONS AND IMPLICATIONS
Oral sCT attenuated diabetic hyperglycaemia in male ZDF rats by improving postprandial glycaemic control, exerting an insulinotropic and glucagonostatic action in the basal state and by preserving pancreatic beta-cell function and beta-cell area.



御礼 誕生日ケーキ・祝賀
1.Barrier-protective integrin αvβ3-IQGAP1-Rac1/CDC42-GTP
M. Bhattacharya, G. Su, X. Su, J. A. Oses-Prieto, J. T. Li, X. Huang, H. Hernandez, A. Atakilit, A. L. Burlingame, M. A. Matthay, and D. Sheppard
IQGAP1 is necessary for pulmonary vascular barrier protection in murine acute lung injury and pneumonia
Am J Physiol Lung Cell Mol Physiol July 1, 2012 303:L12-L19
We recently reported that integrin αvβ3 is necessary for vascular barrier protection in mouse models of acute lung injury and peritonitis. Here, we used mass spectrometric sequencing of integrin complexes to isolate the novel β3-integrin binding partner IQGAP1. Like integrin β3, IQGAP1 localized to the endothelial cell-cell junction after sphingosine-1-phosphate (S1P) treatment, and IQGAP1 knockdown prevented cortical actin formation and barrier enhancement in response to S1P. Furthermore, knockdown of IQGAP1 prevented localization of integrin αvβ3 to the cell-cell junction. Similar to β3-null animals, IQGAP1-null mice had increased pulmonary vascular leak compared with wild-type controls 3 days after intratracheal LPS. In an Escherichia coli pneumonia model, IQGAP1 knockout mice had increased lung weights, lung water, and lung extravascular plasma equivalents of 125I-labeled albumin compared with wild-type controls. Taken together, these experiments indicate that IQGAP1 is necessary for S1P-mediated vascular barrier protection during acute lung injury and is required for junctional localization of the barrier-protective integrin αvβ3
2. ビタミンCで肺血管透過性改善?
Bernard J. Fisher, Donatas Kraskauskas, Erika J. Martin, Daniela Farkas, Jacob A. Wegelin, Donald Brophy, Kevin R. Ward, Norbert F. Voelkel, Alpha A. Fowler III, and Ramesh Natarajan
Mechanisms of attenuation of abdominal sepsis induced acute lung injury by ascorbic acid
Am J Physiol Lung Cell Mol Physiol July 1, 2012 303:L20-L32
Bacterial infections of the lungs and abdomen are among the most common causes of sepsis. Abdominal peritonitis often results in acute lung injury (ALI). Recent reports demonstrate a potential benefit of parenteral vitamin C [ascorbic acid (AscA)] in the pathogenesis of sepsis. Therefore we examined the mechanisms of vitamin C supplementation in the setting of abdominal peritonitis-mediated ALI. We hypothesized that vitamin C supplementation would protect lungs by restoring alveolar epithelial barrier integrity and preventing sepsis-associated coagulopathy. Male C57BL/6 mice were intraperitoneally injected with a fecal stem solution to induce abdominal peritonitis (FIP) 30 min prior to receiving either AscA (200 mg/kg) or dehydroascorbic acid (200 mg/kg). Variables examined included survival, extent of ALI, pulmonary inflammatory markers (myeloperoxidase, chemokines), bronchoalveolar epithelial permeability, alveolar fluid clearance, epithelial ion channel, and pump expression (aquaporin 5, cystic fibrosis transmembrane conductance regulator, epithelial sodium channel, and Na+-K+-ATPase), tight junction protein expression (claudins, occludins, zona occludens), cytoskeletal rearrangements (F-actin polymerization), and coagulation parameters (thromboelastography, pro- and anticoagulants, fibrinolysis mediators) of septic blood. FIP-mediated ALI was characterized by compromised lung epithelial permeability, reduced alveolar fluid clearance, pulmonary inflammation and neutrophil sequestration, coagulation abnormalities, and increased mortality. Parenteral vitamin C infusion protected mice from the deleterious consequences of sepsis by multiple mechanisms, including attenuation of the proinflammatory response, enhancement of epithelial barrier function, increasing alveolar fluid clearance, and prevention of sepsis-associated coagulation abnormalities. Parenteral vitamin C may potentially have a role in the management of sepsis and ALI associated with sepsis.
3. Epithelial-Mesenchymal TransitionにおけるTwist & Smail
Epithelial-Mesenchymal Transition (EMT;上皮間葉移行) は1980年代初めにElizabeth Hayらが提唱した上皮細胞が間葉系様細胞に形態変化する現象であり,初期胚発生における原腸陥入,神経提細胞の運動や器官形成過程特に,心臓や腎臓また口蓋形成での重要性がこれまでに明らかとなっている1。一方,EMTの獲得が運動性の亢進や細胞外基質の蓄積をもたらすことから,癌細胞の浸潤や線維症との関連も示唆されている。TwistはbHLH型転写因子で,myogenin プロモーター上でE12とヘテロ2量体を形成し,遺伝子発現を抑制し,さらにSeathre-Chotzen syndromeの原因遺伝子として知られている。Twistの効果は,細胞特異性や他の補助的分子の存在が必要となる可能性が示唆されている。
Koji Sakamoto, Naozumi Hashimoto, Yasuhiro Kondoh, Kazuyoshi Imaizumi, Daisuke Aoyama, Takashi Kohnoh, Masaaki Kusunose, Motohiro Kimura, Tsutomu Kawabe, Hiroyuki Taniguchi, and Yoshinori Hasegawa
Differential modulation of surfactant protein D under acute and persistent hypoxia in acute lung injury
Am J Physiol Lung Cell Mol Physiol July 1, 2012 303:L43-L53
Hypoxia contributes to the development of fibrosis with epithelial-mesenchymal transition (EMT) via stimulation of hypoxia-inducible factor 1α (HIF-1α) and de novo twist expression. Although hypoxemia is associated with increasing levels of surfactant protein D (SP-D) in acute lung injury (ALI), the longitudinal effects of hypoxia on SP-D expression in lung tissue injury/fibrosis have not been fully evaluated. Here, the involvement of hypoxia and SP-D modulation was evaluated in a model of bleomycin-induced lung injury. We also investigated the molecular mechanisms by which hypoxia might modulate SP-D expression in alveolar cells, by using a doxycycline (Dox)-dependent HIF-1α expression system. Tissue hypoxia and altered SP-D levels were present in bleomycin-induced fibrotic lesions. Acute hypoxia induced SP-D expression, supported by the finding that Dox-induced expression of HIF-1α increased SP-D expression. In contrast, persistent hypoxia repressed SP-D expression coupled with an EMT phenotype and twist expression. Long-term expression of HIF-1α caused SP-D repression with twist expression. Ectopic twist expression repressed SP-D expression. The longitudinal observation of hypoxia and SP-D levels in ALI in vivo was supported by the finding that HIF-1α expression stabilized by acute hypoxia induced increasing SP-D expression in alveolar cells, whereas persistent hypoxia induced de novo twist expression in these cells, causing repression of SP-D and acquisition of an EMT phenotype. Thus this is the first study to demonstrate the molecular mechanisms, in which SP-D expression under acute and persistent hypoxia in acute lung injury might be differentially modulated by stabilized HIF-1α expression and de novo twist expression.
4. Ovalbumin 誘導喘息モデル
Kimitake Tsuchiya, Sana Siddiqui, Paul-André Risse, Nobuaki Hirota, and James G. Martin
The presence of LPS in OVA inhalations affects airway inflammation and AHR but not remodeling in a rodent model of asthma
Am J Physiol Lung Cell Mol Physiol July 1, 2012 303:L54-L63
Ovalbumin (OVA) is the most frequently used allergen in animal models of asthma. Lipopolysaccharide (LPS) contaminating commercial OVA may modulate the evoked airway inflammatory response to OVA. However, the effect of LPS in OVA on airway remodeling, especially airway smooth muscle (ASM) has not been evaluated. We hypothesized that LPS in commercial OVA may enhance allergen-induced airway inflammation and remodeling. Brown Norway rats were sensitized with OVA on day 0. PBS, OVA, or endotoxin-free OVA (Ef-OVA) was instilled intratracheally on days 14, 19, 24. Bronchoalveolar lavage (BAL) fluid, lung, and intrathoracic lymph node tissues were collected 48 h after the last challenge. Immunohistochemistry for α-smooth muscle actin, Periodic-Acid-Schiff staining, and real-time qPCR were performed. Airway hyperresponsiveness (AHR) was also measured. BAL fluid macrophages, eosinophils, neutrophils, and lymphocytes were increased in OVA-challenged animals, and macrophages and neutrophils were significantly lower in Ef-OVA-challenged animals. The ASM area in larger airways was significantly increased in both OVA and Ef-OVA compared with PBS-challenged animals. The mRNA expression of IFN-γ and IL-13 in lung tissues and IL-4 in lymph nodes was significantly increased by both OVA and Ef-OVA compared with PBS and were not significantly different between OVA and Ef-OVA. Monocyte chemoattractant protein (MCP)-1 in BAL fluid and AHR were significantly increased in OVA but not in Ef-OVA. LPS contamination in OVA contributes to the influx of macrophages and MCP-1 increase in the airways and to AHR after OVA challenges but does not affect OVA-induced Th1 and Th2 cytokine expression, goblet cell hyperplasia, and ASM remodeling.
5. 肺高血圧におけるglucose-6-phosphate dehydrogenaseとPKG
Sukrutha Chettimada, Dhwajbahadur K. Rawat, Nupur Dey, Robert Kobelja, Zachary Simms, Michael S. Wolin, Thomas M. Lincoln, and Sachin A. Gupte
Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery
Am J Physiol Lung Cell Mol Physiol July 1, 2012 303:L64-L74
Persistent hypoxic pulmonary vasoconstriction (HPV) plays a significant role in the pathogenesis of pulmonary hypertension, which is an emerging clinical problem around the world. We recently showed that hypoxia-induced activation of glucose-6-phosphate dehydrogenase (Glc-6-PD) in pulmonary artery smooth muscle links metabolic changes within smooth muscle cells to HPV and that inhibition of Glc-6PD reduces acute HPV. Here, we demonstrate that exposing pulmonary arterial rings to hypoxia (20–30 Torr) for 12 h in vitro significantly (P < 0.05) reduces (by 30–50%) SM22α and smooth muscle myosin heavy chain expression and evokes HPV. Glc-6-PD activity was also elevated in hypoxic pulmonary arteries. Inhibition of Glc-6-PD activity prevented the hypoxia-induced reduction in SM22α expression and inhibited HPV by 80–90% (P < 0.05). Furthermore, Glc-6-PD and protein kinase G (PKG) formed a complex in pulmonary artery, and Glc-6-PD inhibition increased PKG-mediated phosphorylation of VASP (p-VASP). In turn, increasing PKG activity upregulated SM22α expression and attenuated HPV evoked by Glc-6-PD inhibition. Increasing passive tension (from 0.8 to 3.0 g) in hypoxic arteries for 12 h reduced Glc-6-PD, increased p-VASP and SM22α levels, and inhibited HPV. The present findings indicate that increases in Glc-6-PD activity influence PKG activity and smooth muscle cell phenotype proteins, all of which affect pulmonary artery contractility and remodeling.
Glc-6-PD and thioredoxin reductase-1 (TxR-1) form a complex with PKG and regulate hypoxia-evoked changes in the expression of contractile proteins in pulmonary artery. Schematic illustrating a potential pathway through which hypoxia affects activity of Glc-6-PD and PKG and expression of contractile proteins. Inhibition of Glc-6-PD-derived NADPH redox reduces TxR-1 activity, which oxidizes thiols on PKG and activates it. Increase in PKG activity reexpresses contractile proteins, whereas myocardin expression is increased in a PKG-independent manner.
総説
Dynamical Systems Approach to Endothelial Heterogeneity
Erzsébet Ravasz Regan and William C. Aird
Circulation Research. 2012;111:110-130
Endothelial cells display remarkable phenotypic heterogeneity. An important goal is to elucidate the scope and mechanisms of endothelial heterogeneity and to use this information to develop vascular bed–specific therapies. We reexamine our current understanding of the molecular basis of endothelial heterogeneity. We introduce multistability as a new explanatory framework in vascular biology. We draw on the field of nonlinear dynamics to propose a dynamical systems framework for modeling multistability and its derivative properties, including robustness, memory, and plasticity. Our perspective allows for both a conceptual and quantitative description of system-level features of endothelial regulation.
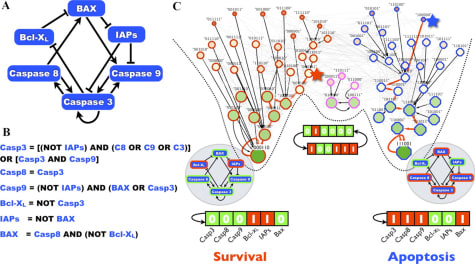
Boolean model of a caspase-mediated survival-apoptosis switch
血管内皮細胞の細胞内情報伝達を良くまとめている総説です。ダイナミックセオリーも,その通りというところがたくさんあります。
Agonist-mediated signaling pathways with cross-talk
原著論文
1. Inefficient Reprogramming of Fibroblasts into Cardiomyocytes Using Gata4, Mef2c, and Tbx5
Jenny X. Chen, Markus Krane, Marcus-Andre Deutsch, Li Wang, Moshe Rav-Acha, Serge Gregoire, Marc C. Engels, Kuppusamy Rajarajan, Ravi Karra, E. Dale Abel, Joe C. Wu, David Milan, and Sean M. Wu
Circulation Research. 2012;111:50-55
Rationale: Direct reprogramming of fibroblasts into cardiomyocytes is a novel strategy for cardiac regeneration. However, the key determinants involved in this process are unknown.
Objective: To assess the efficiency of direct fibroblast reprogramming via viral overexpression of GATA4, Mef2c, and Tbx5 (GMT).
Methods and Results: We induced GMT overexpression in murine tail tip fibroblasts (TTFs) and cardiac fibroblasts (CFs) from multiple lines of transgenic mice carrying different cardiomyocyte lineage reporters. We found that the induction of GMT overexpression in TTFs and CFs is inefficient at inducing molecular and electrophysiological phenotypes of mature cardiomyocytes. In addition, transplantation of GMT infected CFs into injured mouse hearts resulted in decreased cell survival with minimal induction of cardiomyocyte genes.
Conclusions: Significant challenges remain in our ability to convert fibroblasts into cardiomyocyte-like cells and a greater understanding of cardiovascular epigenetics is needed to increase the translational potential of this strategy.
2. コロニンsiRNA
Role of Coronin 1B in PDGF-Induced Migration of Vascular Smooth Muscle Cells
Holly C. Williams, Alejandra San Martín, Candace M. Adamo, Bonnie Seidel-Rogol, Lily Pounkova, Srinivasan Raju Datla, Bernard Lassègue, James E. Bear, and Kathy Griendling
Circulation Research. 2012;111:56-65
Rationale: The type I subclass of coronins, a family of actin-binding proteins, regulates various actin-dependent cellular processes, including migration. However, the existence and role of coronins in vascular smooth muscle cell (VSMC) migration has yet to be determined.
Objective: The goal of the present study was to define the mechanism by which coronins regulate platelet-derived growth factor (PDGF)–induced VSMC migration.
Methods and Results: Coronin 1B (Coro1B) and 1C (Coro1C) were both found to be expressed in VSMCs at the mRNA and protein levels. Downregulation of Coro1B by siRNA increases PDGF-induced migration, while downregulation of Coro1C has no effect. We confirmed through kymograph analysis that the Coro1B-downregulation-mediated increase in migration is directly linked to increased lamellipodial protraction rate and protrusion distance in VSMC. In other cell types, coronins exert their effects on lamellipodia dynamics by an inhibitory interaction with the ARP2/3 complex, which is disrupted by the phosphorylation of Coro1B. We found that PDGF induces phosphorylation of Coro1B on serine-2 via PKCɛ, leading to a decrease in the interaction of Coro1B with the ARP2/3 complex. VSMCs transfected with a phosphodeficient S2A Coro1B mutant showed decreased migration in response to PDGF, suggesting that the phosphorylation of Coro1B is required for the promotion of migration by PDGF. In both the rat and mouse, Coro1B phosphorylation was increased in response to vessel injury in vivo.
Conclusions: Our data suggest that phosphorylation of Coro1B and the subsequent reduced interaction with ARP2/3·complex participate in PDGF-induced VSMC migration, an important step in vascular lesion formation.
血管平滑筋には,Coronin 1BとICが存在する。
RT-PCR Primer
Coro1B (upstream primer, 5′-ACA TGT CCT TCC GAA AAG TTG TGC-3′; downstream primer, 5′-CTG ATC CAC TGG CAA TGA CTT CGT-3′);Coro1C (upstream primer, 5′-TGT CTT CAC TAC TGG TTT TAG CCG TA-3′; downstream primer, 5′-TCT AGC TTT GAA ATG CGC TCG TCT-3′); and GAPDH (upstream primer, 5′-AAT GGG GTG ATG CTG GTG CTG AGT A-3′; downstream primer, 5′-GGA AGA ATG GGA GTT GCT GTT GAA G-3′)
siRNA
Coro1B (5′-CAG CAC CTT CTG CGC AGT CAA-3′), Coro1c (5′-ACG AGA GAA AGT GTG AAC CTA-3′) and PKCɛ (5′-CCC GGG AAG AGC CAA TAC TTA-3′)
コロニンについて
Coronins are a family of evolutionarily conserved tryptophan (W) and aspartic acid (D)-repeat actin-binding proteins known to control a variety of cellular processes involving actin dynamics.The coronin protein family includes 7 proteins in mammals, separated into 3 subclasses (types I, II, and III) based on phylogenetic similarity.The type I coronins consist of coronin 1A (Coro1A), 1B (Coro1B), and 1C (Coro1C) and are the most studied coronin subfamily. Coro1A is highly expressed in cells of hematopoietic lineage in addition to various tissues of the nervous system, while having significantly lower expression in other tissues of the body.1On the other hand, Coro1B and Coro1C are more ubiquitously expressed at higher levels in most tissues.11 Coro1B localizes to the plasma membrane, regulates lamellipodia formation, and when phosphorylated is no longer able to bind ARP2/3 and inhibit its actin nucleation abilities. In contrast, in epithelial cells, Coro1C was demonstrated to regulate focal adhesion dynamics and increase wound closure. Thus, Coro1B and 1C can potentially influence migration via several different mechanisms, and their overall impact is likely to be a function of the complement of coronins expressed in a given cell type.
3. リアノジン受容体を介したCICR
Ryanodine Receptor Current Amplitude Controls Ca2+ Sparks in Cardiac Muscle
Tao Guo, Dirk Gillespie, and Michael Fill
Circulation Research. 2012;111:28-36
Rationale: In cardiac muscle, Ca2+-induced Ca2+ release (CICR) from the sarcoplasmic reticulum (SR) is mediated by ryanodine receptor (RyR) Ca2+ release channels. The inherent positive feedback of CICR is normally well-controlled. Understanding this control mechanism is a priority because its malfunction has life-threatening consequences.
Objective: We show that CICR local control is governed by SR Ca2+ load, largely because load determines the single RyR current amplitude that drives inter-RyR CICR.
Methods and Results: We differentially manipulated single RyR Ca2+ flux amplitude and SR Ca2+ load in permeabilized ventricular myocytes as an endogenous cell biology model of the heart. Large RyR-permeable organic cations were used to interfere with Ca2+ conductance through the open RyR pore. Single-channel studies show this attenuates current amplitude without altering other aspects of RyR function. In cells, the same experimental maneuver increased resting SR Ca2+ load. Despite the increased load, Ca2+ spark (inter-RyR CICR events) frequency decreased and sparks terminated earlier.
Conclusions: Spark local control follows single RyR current amplitude, not simply SR Ca2+ load. Spark frequency increases with load because spontaneous RyR openings at high loads produce larger currents (ie, a larger CICR trigger signal). Sparks terminate when load falls to the point at which single RyR current amplitude is no longer sufficient to sustain inter-RyR CICR. Thus, RyRs that spontaneously close no longer reopen and local Ca2+ release ends.
付録:TAM Receptor
Nature Reviews Immunology 8, 327-336 (May 2008)
Immunobiology of the TAM receptors
Recent studies have revealed that the TAM receptor protein tyrosine kinases ― TYRO3, AXL and MER ― have pivotal roles in innate immunity. They inhibit inflammation in dendritic cells and macrophages, promote the phagocytosis of apoptotic cells and membranous organelles, and stimulate the maturation of natural killer cells. Each of these phenomena may depend on a cooperative interaction between TAM receptor and cytokine receptor signalling systems. Although its importance was previously unrecognized, TAM signalling promises to have an increasingly prominent role in studies of innate immune regulation.
TYRO3 (also known as BRT, DTK, RSE, SKY and TIF), AXL (also known as ARK, TYRO7 and UFO) and MER (also known as EYK, NYM and TYRO12) are receptor protein tyrosine kinases (PTKs) that are expressed by dendritic cells, macrophages and immature natural killer (NK) cells of the immune system, Sertoli cells of the testis, retinal pigment epithelial (RPE) cells of the eye, and several other cell types. TAM receptor dimers bind to their two ligands, growth-arrest-specific 6 (GAS6) and protein S, through interaction between the two N-terminal immunoglobulin-like domains of the receptors and the two C-terminal laminin G (LG) regions, which together make up the SHBG (sex hormone binding globulin) domain, of the ligands. (The solved X-ray crystal structure of the GAS6 SHBG domain bound to the immunoglobulin domains of AXL7 reveals that both ligand and receptor crystallize as dimers.) Via their N-terminal Gla domains, GAS6 and protein S then bind to phosphatidylserine that is displayed on the extracellular surface of the plasma membranes of apoptotic cells or on the outer segments of photoreceptors. EGF, epidermal growth factor; FNIII, fibronectin type III.
Editorial Focus
Jeffrey E. Gotts and Michael A. Matthay
Mesenchymal stem cells and the stem cell niche: a new chapter
Am J Physiol Lung Cell Mol Physiol June 1, 2012 302:L1147-L1149

BONE MARROW-DERIVED mesenchymal stem/stromal cells (MSCs) are self-renewing multipotent cells with therapeutic effects in diverse models of tissue injury. In the rodent lung, MSCs reduce collagen deposition in the bleomycin model of pulmonary fibrosis and reduce lung injury and improve survival following intrapulmonary delivery of endotoxin or Escherichia coli and following severe gram-negative peritonitis. In the hyperoxia model of bronchopulmonary dysplasia, exposure to high concentrations of oxygen during early postnatal life in rats and mice causes simplification of alveolar and lung capillary structure and reduced pulmonary capillary surface area, leading to pulmonary hypertension. Two groups reported simultaneously in 2009 that MSCs given by airway to rats or by blood to mice during prolonged hyperoxia in early postnatal life prevented arrested alveolar growth. However, engraftment of MSCs during hyperoxia and in other models has not accounted for the therapeutic effects, thus prompting a search for other mechanisms. MSCs are potent immunomodulators, suppressing several functions of lymphocytes, natural killer cells, and monocytes, and reduce inflammatory cell lung infiltrates and cytokines during sepsis and acute lung injury. In addition, MSCs have direct antibacterial effects, secrete epithelial growth factors, and can rescue epithelial cellular bioenergetics with mitochondrial transfer.
Original Articles
■ β-Adrenergic agonists differentially regulate highly selective and nonselective epithelial sodium channels to promote alveolar fluid clearance in vivo
Charles A. Downs, Lisa H. Kriener, Ling Yu, Douglas C. Eaton, Lucky Jain, and My N. Helms
Am J Physiol Lung Cell Mol Physiol June 1, 2012 302:L1167-L1178
ENaCの活性化は,β1受容体でもβ2受容体刺激でも生じ,肺の水クリアランスを改善する。
デノパミン β1-stimulation
タブタリン β2-stimulation
β-Adrenergic receptors (β-AR) increase epithelial sodium channel (ENaC) activity to promote lung fluid clearance. However, the effect of selective β-AR agonist on highly selective cation (HSC) channels or nonselective cation (NSC) channels in alveolar type 1 (T1) and type 2 (T2) cells is unknown. We hypothesized that stimulation with β(1)-AR agonist (denopamine) or β(2)-AR agonist (terbutaline) would increase HSC and/or NSC channel activity in alveolar epithelial cells. We performed single-channel measurements from T1 and T2 cells accessed from rat lung slices. Terbutaline (20 μM) increased HSC ENaC activity (open probability, NP(o)) in T1 (from 0.96 ± 0.61 to 1.25 ± 0.71, n = 5, P
■ The p110δ subunit of PI3K regulates bone marrow-derived eosinophil trafficking and airway eosinophilia in allergen-challenged mice
Bit Na Kang, Sung Gil Ha, Xiao Na Ge, M. Reza Hosseinkhani, Nooshin S. Bahaie, Yana Greenberg, Malcolm N. Blumenthal, Kamal D. Puri, Savita P. Rao, and P. Sriramarao
Am J Physiol Lung Cell Mol Physiol June 1, 2012 302:L1179-L1191
好酸球のリクルートメントとトラフィッキング
CD11b・・MAC1
CD49・・α4
CRA:cockroach antigenでのマウスの気道過敏性に関するin vivo研究も併設
Trafficking and recruitment of eosinophils during allergic airway inflammation is mediated by the phosphatidylinositol 3-kinase (PI3K) family of signaling molecules. The role played by the p110δ subunit of PI3K (PI3K p110δ) in regulating eosinophil trafficking and recruitment was investigated using a selective pharmacological inhibitor (IC87114). Treatment with the PI3K p110δ inhibitor significantly reduced murine bone marrow-derived eosinophil (BM-Eos) adhesion to VCAM-1 as well as ICAM-1 and inhibited activation-induced changes in cell morphology associated with reduced Mac-1 expression and aberrant cell surface localization/distribution of Mac-1 and α4. Infused BM-Eos demonstrated significantly decreased rolling and adhesion in inflamed cremaster muscle microvessels of mice treated with IC87114 compared with vehicle-treated mice. Furthermore, inhibition of PI3K p110δ significantly attenuated eotaxin-1-induced BM-Eos migration and prevented eotaxin-1-induced changes in the cytoskeleton and cell morphology. Knockdown of PI3K p110δ with siRNA in BM-Eos resulted in reduced rolling, adhesion, and migration, as well as inhibition of activation-induced changes in cell morphology, validating its role in regulating trafficking and migration. Finally, in a mouse model of cockroach antigen-induced allergic airway inflammation, oral administration of the PI3K p110δ inhibitor significantly inhibited airway eosinophil recruitment, resulting in attenuation of airway hyperresponsiveness in response to methacholine, reduced mucus secretion, and expression of proinflammatory molecules (found in inflammatory zone-1 and intelectin-1). Overall, these findings indicate the important role played by PI3K p110δ in mediating BM-Eos trafficking and migration by regulating adhesion molecule expression and localization/distribution as well as promoting changes in cell morphology that favor recruitment during inflammation.
■ RAGE35
RAGE signaling by alveolar macrophages influences tobacco smoke-induced inflammation
Adam B. Robinson, KacyAnn D. Johnson, Brock G. Bennion, and Paul R. Reynolds
Am J Physiol Lung Cell Mol Physiol June 1, 2012 302:L1192-L1199
CSE・・タバコの煙暴露モデル:肺胞マクロファージのRAGEが増加する
cigarette smoke extractはRAGEのリガンドか,RAGEリガンドを産生する可能性がある。
RAGE→RAS→P38MAPK ?
RAGE-KOモデルの使用
Receptors for advanced glycation end-products (RAGE) are multiligand cell surface receptors of the immunoglobin family expressed by epithelium and macrophages, and expression increases following exposure to cigarette smoke extract (CSE). The present study sought to characterize the proinflammatory contributions of RAGE expressed by alveolar macrophages (AMs) following CSE exposure. Acute exposure of mice to CSE via nasal instillation revealed diminished bronchoalveolar lavage (BAL) cellularity and fewer AMs in RAGE knockout (KO) mice compared with controls. Primary AMs were obtained from BAL, exposed to CSE in vitro, and analyzed. CSE significantly increased RAGE expression by wild-type AMs. Employing ELISAs, wild-type AMs exposed to CSE had increased levels of active Ras, a small GTPase that perpetuates proinflammatory signaling. Conversely, RAGE KO AMs had less Ras activation compared with wild-type AMs after exposure to CSE. In RAGE KO AMs, assessment of p38 MAPK and NF-κB, important intracellular signaling intermediates induced during an inflammatory response, revealed that CSE-induced inflammation may occur in part via RAGE signaling. Lastly, quantitative RT-PCR revealed that the expression of proinflammatory cytokines including TNF-α and IL-1β were detectably decreased in RAGE KO AMs exposed to CSE compared with CSE-exposed wild-type AMs. These results reveal that primary AMs orchestrate CSE-induced inflammation, at least in part, via RAGE-mediated mechanisms.
■ Platelets induce endothelial tissue factor expression in a mouse model of acid-induced lung injury
Memet T. Emin, Li Sun, Alice Huertas, Shonit Das, Jahar Bhattacharya, and Sunita Bhattacharya
Am J Physiol Lung Cell Mol Physiol June 1, 2012 302:L1209-L1220
Dept. of Pediatrics, Columbia Univ., 630 W. 168th St., BB 8-812, New York, NY 10032. sb80@columbia.edu.
Although the lung expresses procoagulant proteins under inflammatory conditions, underlying mechanisms remain unclear. Here, we addressed lung endothelial expression of tissue factor (TF), which initiates the coagulation cascade and expression of which signifies development of a procoagulant phenotype in the vasculature. To establish the model of acid-induced acute lung injury (ALI), we intranasally instilled anesthetized mice with saline or acid. Then 2 h later, we isolated pulmonary vascular cells for flow cytometry and confocal microscopy to detect the leukocyte antigen, CD45 and the endothelial markers VE-cadherin and von Willebrand factor (vWf). Acid increased both the number of vWf-expressing cells as well as TF and P-selectin expressions on these cells. All of these effects were markedly inhibited by treating mice with antiplatelet serum, suggesting the involvement of platelets. The increased expressions of TF, vWf, and P-selectin in response to acid also occurred in platelets. Moreover, the effects were replicated in endothelial cells derived from isolated, blood-perfused lungs. However, the effect was inhibited completely in lungs perfused with platelet-depleted and, to a lesser extent, with leukocyte-depleted blood. Acid injury increased endothelial expressions of the platelet proteins, CD41 and CD42b, providing evidence that platelet proteins were transferred to the vascular surface. Reactive oxygen species (ROS) were implicated in these responses, in that the endothelial and platelet protein expressions were inhibited. We conclude that acid-induced ALI causes NOX2-mediated ROS generation that activates platelets, which then generate a procoagulant endothelial surface.
■ Interaction with CREB binding protein modulates the activities of Nrf2 and NF-κB in cystic fibrosis airway epithelial cells
Assem G. Ziady, Andrew Sokolow, Samuel Shank, Deborah Corey, Ross Myers, Scott Plafker, and Thomas J. Kelley
Am J Physiol Lung Cell Mol Physiol June 1, 2012 302:L1221-L1231
cAMP→CREB→NRF2活性
cAMP活性によるNRF2活性vsNF-κB活性の調節作用
NRF2はKeap1で制御されており,25個のシステインのうち,Cys151,Cys273,Cys288などが酸化修飾を受け,NRF2のユビキチン化が減少し,NRF2活性が持続することにより,抗酸化物質の転写が亢進する。
Cystic fibrosis (CF) is characterized by inflammatory lung disease that significantly contributes to morbidity and mortality. Airway epithelial cells play a role in the inflammatory signaling in CF and have been reported to exhibit a number of dysfunctions in signaling cascades that modulate inflammation. Previously, we reported that the activity of nuclear factor erythroid-derived-like 2 (Nrf2), a transcription factor that regulates antioxidant and cytoprotective protein expression, is diminished in CF epithelia (7). In this report, we examined the mechanism of Nrf2 dysregulation in vitro in human airway epithelial cell lines and primary cells and in vivo in nasal epithelia excised from ΔF508 CF mutant mice. We found that cAMP-mediated signaling markedly reduces Nrf2 activity in CF vs. non-CF cells. Rp-cAMPS, a cAMP competitor, significantly corrected Nrf2 activity in CF cells, predominantly by increasing the nuclear accumulation of the transcription factor. Furthermore, we found that Rp-cAMPS significantly decreased NF-κB activation following inflammatory stimulation of CF cells. Further investigation revealed that Nrf2 and NF-κB compete for the transcriptional coactivator cAMP responsive element-binding protein (CREB) binding protein (CBP) and that Rp-cAMPS shifts CBP association in favor of Nrf2. Thus our findings provide a link between feedback to CF transmembrane regulator dysfunction and dysregulation of an inflammatory signaling pathway that modulates the coordinated activities of Nrf2 and NF-κB. Furthermore, our studies suggest that strategies that shift CBP association away from NF-κB and toward Nrf2 could have potential therapeutic efficacy for reducing inflammation in patients with CF.
Eliot S. Katz, Ron B. Mitchell, and Carolyn M. D'Ambrosio.Am. J. Respir. Crit. Care Med. 2012; 185: 805-816.
Obstructive sleep apnea in infants has a distinctive pathophysiology, natural history, and treatment compared with that of older children and adults. Infants have both anatomical and physiological predispositions toward airway obstruction and gas exchange abnormalities; including a superiorly placed larynx, increased chest wall compliance, ventilation–perfusion mismatching, and ventilatory control instability. Congenital abnormalities of the airway, such as laryngomalacia, hemangiomas, pyriform aperture stenosis, choanal atresia, and laryngeal webs, may also have adverse effects on airway patency. Additional exacerbating factors predisposing infants toward airway collapse include neck flexion, airway secretions, gastroesophageal reflux, and sleep deprivation. Obstructive sleep apnea in infants has been associated with failure to thrive, behavioral deficits, and sudden infant death. The proper interpretation of infant polysomnography requires an understanding of normative data related to gestation and postconceptual age for apnea, arousal, and oxygenation. Direct visualization of the upper airway is an important diagnostic modality in infants with obstructive apnea. Treatment options for infant obstructive sleep apnea are predicated on the underlying etiology, including supraglottoplasty for severe laryngomalacia, mandibular distraction for micrognathia, tonsillectomy and/or adenoidectomy, choanal atresia repair, and/or treatment of gastroesophageal reflux.
2. 原著 A Critical Role for Muscle Ring Finger-1 in Acute Lung Injury–associated Skeletal Muscle Wasting
D. Clark Files, Franco R. D'Alessio, Laura F. Johnston, Priya Kesari, Neil R. Aggarwal, Brian T. Garibaldi, Jason R. Mock, Jessica L. Simmers, Antonio DeGorordo, Jared Murdoch, Monte S. Willis, Cam Patterson, Clarke G. Tankersley, Maria L. Messi, Chun Liu, Osvaldo Delbono, J. David Furlow, Sue C. Bodine, Ronald D. Cohn, Landon S. King, and Michael T. Crow. Am. J. Respir. Crit. Care Med. 2012; 185: 825-834.
ALIにおける骨格筋萎縮に関する原著 ポイント:Atrogin-1とMuscle ring finger-1(MuRF-1)のシグナル知識を整理すること
Skeletal muscle weakness is a common finding not only among patients with ALI, but also in patients with other critical illnesses. Clinically apparent weakness is present in 20–50% of patients with critical illness and has been shown to be an independent risk factor for mortality in these patients. A variety of terms have been used in the literature to describe the myopathic weakness in these patients including acute quadriplegic myopathy, critical illness myopathy, and thick filament myopathy, the latter referring to the preferential loss of myosin observed in the muscles of these patients (1–3).
1.al-Lozi MT, Pestronk A, Yee WC, Flaris N, Cooper J. Rapidly evolving myopathy with myosin-deficient muscle fibers. Ann Neurol 1994;35:273–279. CrossRefMedline
2. Norman H, Zackrisson H, Hedstrom Y, Andersson P, Nordquist J, Eriksson LI, Libelius R, Larsson L. Myofibrillar protein and gene expression in acute quadriplegic myopathy. J Neurol Sci 2009;285:28–38. CrossRefMedline
3.↵ Sher JH, Shafiq SA, Schutta HS. Acute myopathy with selective lysis of myosin filaments. Neurology 1979;29:100–106.
Their data demonstrate that ALI in mice produces marked skeletal muscle wasting and dysfunction similar to that observed in patients with ALI. Muscle wasting and dysfunction in this model is associated with markedly increased NF-κB activity and MuRF1 transcriptional activation and is suppressed by genetic inactivation or biochemical suppression of MuRF1. In contrast, muscle wasting in PF mice is not affected by suppressing MuRF1. It remains to be determined if MuRF1 is expressed in the skeletal muscles of humans with ALI and whether blockade of MuRF1 could prevent muscle wasting in these patients. Although recognizing that the results obtained in the mouse may have limited relevance to ALI in humans, MuRF1 seems to be an attractive therapeutic target for ALI-associated skeletal muscle wasting.
Studies over the last 12 years have defined important roles for muscle-specific genes that regulate muscle wasting in well-defined models of skeletal muscle atrophy, including immobilization, denervation, and hindlimb suspension (4). Prominent among these are the genes Fbx032 (atrogin1 or MAFbx) and Trim63 (muscle ring finger protein-1 [MuRF1]), both of which function as ubiquitin E3 ligases in the proteasome-mediated degradation of skeletal muscle proteins (4, 5). Up-regulation of MuRF1 and atrogin has also been observed in the peripheral muscles of patients with chronic obstructive pulmonary disease and in the diaphragms of mechanically ventilated brain-dead patients (6, 7).
4. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704–1708.
5. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle- specific f-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 2001;98:14440–14445.
6. Doucet M, Russell A, Léger B, Debigaré R, Joanisse DR, Caron M, LeBlanc P, Maltais F. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007;176:261–269.
7. Hussain SN, Mofarrahi M, Sigala I, Kim HC, Vassilakopoulos T, Maltais F, Bellenis I, Chaturvedi R, Gottfried SB, Metrakos P, et al. Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am J Respir Crit Care Med 2010;182:1377–1386.
Rationale: Acute lung injury (ALI) is a debilitating condition associated with severe skeletal muscle weakness that persists in humans long after lung injury has resolved. The molecular mechanisms underlying this condition are unknown.
Objectives: To identify the muscle-specific molecular mechanisms responsible for muscle wasting in a mouse model of ALI.
Methods: Changes in skeletal muscle weight, fiber size, in vivo contractile performance, and expression of mRNAs and proteins encoding muscle atrophy–associated genes for muscle ring finger-1 (MuRF1) and atrogin1 were measured. Genetic inactivation of MuRF1 or electroporation-mediated transduction of miRNA-based short hairpin RNAs targeting either MuRF1 or atrogin1 were used to identify their role in ALI-associated skeletal muscle wasting.
Measurements and Main Results: Mice with ALI developed profound muscle atrophy and preferential loss of muscle contractile proteins associated with reduced muscle function in vivo. Although mRNA expression of the muscle-specific ubiquitin ligases, MuRF1 and atrogin1, was increased in ALI mice, only MuRF1 protein levels were up-regulated. Consistent with these changes, suppression of MuRF1 by genetic or biochemical approaches prevented muscle fiber atrophy, whereas suppression of atrogin1 expression was without effect. Despite resolution of lung injury and down-regulation of MuRF1 and atrogin1, force generation in ALI mice remained suppressed.
Conclusions: These data show that MuRF1 is responsible for mediating muscle atrophy that occurs during the period of active lung injury in ALI mice and that, as in humans, skeletal muscle dysfunction persists despite resolution of lung injury.
3. 原著 Activation of Mitochondrial Biogenesis by Heme Oxygenase-1–mediated NF-E2–related Factor-2 Induction Rescues Mice from Lethal Staphylococcus aureus Sepsis
Nancy Chou MacGarvey, Hagir B. Suliman, Raquel R. Bartz, Ping Fu, Crystal M. Withers, Karen E. Welty-Wolf, and Claude A. Piantadosi.Am. J. Respir. Crit. Care Med. 2012; 185: 851-861.
ポイント:AKt-1 KOマウスの使用,Nrf2 KOマウスの使用,S.aureus生菌敗血症モデル,His3 as a nuclear reference protein
Studies were preapproved by our Institutional Animal Care and Use Committee. C57Bl6/J (WT) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Nrf2-/- mice (Riken, Saitama, Japan) and Akt1-/- mice (Jackson) were bred institutionally and both sexes used at 9–15 weeks of age.
In C57BL/6J mice (WT), sepsis induced by implanting fibrin clots containing live 5 × 107 cfu S. aureus into the peritoneum followed by fluid resuscitation produces dose-dependent organ damage and lethality .
Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med 2007;176:768–777.
Hmox1 is thought to protect against sepsis-induced tissue damage, and it is induced by multiple transcriptional elements that respond to inflammation, especially the basic leucine zipper transcription factor, NF-E2 related factor-2 (Nrf2) (1). Nrf2 is normally sequestered in the cytosol by the cysteine-rich Kelch-like ECH-associated protein 1 (2), and Kelch-like ECH-associated protein 1 oxidation allows Nrf2 nuclear translocation (3) and binding to antioxidant response element (ARE) motifs located 5′ to the Hmox-1 transcription start site (4). Nrf2 also occupies activating ARE motifs in the nuclear respiratory factor-1 (NRF-1) promoter, and under the influence of CO, Nrf2 and NRF-1 along with NRF-2 (Gabpa) and the peroxisome proliferator-activated receptor gamma coactivator (PGC)-1 coactivators stimulate mitochondrial biogenesis (5). Nrf2 also influences the innate immune response and survival in cecal ligation and puncture (6) and modulates leukocyte function in sepsis (7).
1.↵ Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a cap'n'collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 1999;274:26071–26078. Abstract/FREE Full Text
2↵ Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13:76–86. Abstract/FREE Full Text
3.↵ Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA 2002;99:11908–11913. Abstract/FREE Full Text
4.↵ Alam J, Igarashi K, Immenschuh S, Shibahara S, Tyrrell RM. Regulation of heme oxygenase-1 gene transcription: Recent advances and highlights from the international conference (Uppsala, 2003) on heme oxygenase. Antioxid Redox Signal 2004;6:924–933. Medline
5.↵ Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 2011;1813:1269–1278. CrossRefMedline
6.↵ Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 2006;116:984–995. CrossRefMedline
7.↵ Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med 2011;184:928–938.
4. To the Editor: 敗血症病態におけるアドレナリン作動性β-受容体活性について
Michael Eisenhut
Inflammation-induced Desensitization of β-Receptors in Acute Lung Injury
Am. J. Respir. Crit. Care Med. 2012; 185: 894
The authors of a randomized controlled trial of the inhaled β-agonist albuterol in patients with acute lung injury mentioned as reasons for a failure to improve outcome poor delivery, damage to alveolar epithelium, down-regulation of β2-receptors, possible differences in genetic variants of the β-receptor in the groups of the trial, and the possibility that fluid clearance was already maximized by lung-protective ventilation and a fluid-conservative hemodynamic strategy (1). The authors did not consider an important phenomenon observed in patients with severe systemic inflammatory response syndromes, who constituted the majority in this trial. It is a reduced responsiveness of β-receptors to β-agonist induced by inflammatory mediators.
Regarding this reduced responsiveness of β-receptors in a systemic inflammatory response, most can be learned from previous research into induction of β2-receptor hyporesponsiveness in models of asthma and airway smooth muscle cells. One mechanism is desensitization by cytokines including nterleukin-1β (IL-1), tumor necrosis factor, transforming growth factor-β, and IL-13 (2). In human airway smooth muscle cells, IL-1 induces cyclooxygenase-2 (COX-2) expression and hence prostaglandin E2 (PGE2). PGE2 causes cAMP formation, and it has been shown that protein kinase A (PKA) activated by cAMP can phosphorylate the β2-adrenoceptor and induce desensitization of the receptor by interference with its attachment with G protein (3). PKA has also been shown to down-regulate β2-receptors by inhibition of transcription and was found to activate phosphodiesterase-4, which reduces cAMP levels (4). IL-1–mediated activation of the Gi pathway by up-regulation of inhibitory G proteins Giα1, Giα2, and Giα3 causes uncoupling of the β-adrenergic receptors from the adenylate cyclase (5).
Another avenue for rapid desensitization of the β2-receptor has been discovered in a rat model, where IL-1 elevated intracellular G protein–coupled receptor kinase-2 (GRK2), an enzyme that was detected in rat alveolar epithelial cells (6) and is the key enzyme in rapid desensitization of β2-receptors to endogenous and exogenous catecholamines.
Dexamethasone has been shown to prevent this IL-1–induced up-regulation of GRK-2 levels (5). Dexamethasone also inhibits IL-1β–induced COX-2 expression and PGE2 release (3).
Future research into pathways to improve the outcome of lung injury with β-agonists needs to explore the regulation of β-agonist sensitivity in human alveolar epithelial cells in vitro. β-Agonists may improve outcome of lung injury if their receptors are (hyper-) sensitized by systemic or local application of corticosteroids.
1.↵ The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Randomized, placebo-controlled clinical trial of an aerosolized beta 2-agonist for treatment of acute lung injury. Am J Respir Crit Care Med 2011;184:561–568. Abstract/FREE Full Text
2.↵ Shore SA. Cytokine regulation of beta-adrenergic responses in airway smooth muscle. J Allergy Clin Immunol 2002;110:255–260. CrossRefMedline
3.↵ Laporte JD, Moore PE, Panettieri RA, Moeller W, Heyder J, Shore SA. Prostanoids mediate IL-1beta-induced beta-adrenergic hyporesponsiveness in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 1998;275:L491–L501. Abstract/FREE Full Text
4.↵ Guo M, Pascual RM, Wang S, Fontana MF, Valancius CA, Panettieri RA, Tilley SL, Penn RB. Cytokines regulate beta-2-adrenergic receptor responsiveness in airway smooth muscle via multiple PKA-and EP2 receptor-dependent mechanisms. Biochemistry 2005;44:13771–13782. CrossRefMedline
5.↵ Mak JCW, Hisada T, Salmon M, Barnes PJ, Chung KF. Glucocorticoids reverse IL-1 beta-induced impairment of beta-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinases. Br J Pharmacol 2002;135:987–996. CrossRefMedline
6.↵ Liebler JM, Borok Z, Li X, Zhou B, Sandoval AJ, Kim K-J, Crandall ED. Alveolar epithelial type I cells express beta 2-adrenergic receptors and G-protein receptor kinase 2. J Histochem Cytochem 2004;52:759–767.
Michael A. Matthay, B. Taylor Thompson, and Roy Brower
Inflammation-induced Desensitization of β-Receptors in Acute Lung Injury
Am. J. Respir. Crit. Care Med. 2012; 185: 894-895
We appreciate the thoughtful comments by Dr. Eisenhut regarding potential additional explanations for why the aerosolized β2-agonist albuterol was not effective in improving clinical outcomes in our phase III clinical trial in patients with acute lung injury (1). We agree that proinflammatory substances such as IL-1 may decrease the capacity of alveolar epithelial cells to clear alveolar edema fluid (2). Dr. Eisenhut notes that there is experimental evidence that dexamethasone might be effective in preventing IL-1–dependent desensitization of β2-receptors to catecholamines. Further, he proposes that β-agonists might improve the outcome of lung injury if the receptors were hypersensitized by systemic or local application of corticosteroids.
A rigorous assessment of combined β-agonist and corticosteroid therapy would require a careful pre-clinical assessment followed by well-designed clinical trials. The initial clinical studies would need a strong focus on safety, especially because of the recently published BALTI-2 trial with intravenous albuterol, indicating harmful effects with β-agonist monotherapy (3). In addition, assays of glucocorticoid responsiveness demonstrate considerable variability among normal individuals and among patients with various clinical diseases. There are several explanations for this variability; some of it may be attributed to genetic variations in the glucocorticoid receptor (4), but some of the variability may be attributable to the effects of the inflammatory environment (5–8).
1.↵ National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Randomized, placebo-controlled clinical trial of an aerosolized beta-agonist for treatment of acute lung injury. Am J Respir Crit Care Med 2011;184:561–568. Abstract/FREE Full Text
2.↵ Folkesson HG, Matthay MA. Alveolar epithelial ion and fluid transport: recent progress. Am J Respir Cell Mol Biol 2006;35:10–19. FREE Full Text
3.↵ Gao F, Perkins GD, Gates S, Young D, McAuley D, Tunnicliffe W, Khan Z, Lamb SE. Effect of intravenous β-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet 2012;379:229–235. CrossRefMedline
4.↵ Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, Lange C, Lazarus R, Sylvia J, Klanderman B, et al. Genomewide association between glcci1 and response to glucocorticoid therapy in asthma. N Engl J Med 2011;365:1173–1183. CrossRefMedline
5.↵ Siebig S, Meinel A, Rogler G, Klebl E, Wrede CE, Gelbmann C, Froh S, Rockmann F, Bruennler T, Schoelmerich J, et al. Decreased cytosolic glucocorticoid receptor levels in critically ill patients. Anaesth Intensive Care 2010;38:133–140. Medline
6. Kam JC, Szefler SJ, Surs W, Sher ER, Leung DY. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and t cell response to glucocorticoids. J Immunol 1993;151:3460–3466. Abstract
7. Spahn JD, Szefler SJ, Surs W, Doherty DE, Nimmagadda SR, Leung DY. A novel action of IL-13: induction of diminished monocyte glucocorticoid receptor-binding affinity. J Immunol 1996;157:2654–2659. Abstract
8.↵ Meduri GU, Yates CR. Systemic inflammation-associated glucocorticoid resistance and outcome of ARDS. Ann N Y Acad Sci 2004;1024:24–53. CrossRefMedline
EPAC(exchange protein activated by cAMP)も最近は極めて有名な細胞内情報伝達蛋白となりました。
名大救急・集中治療ジャーナルクラブ ~毎週木曜日開催中~
総説
■ Charlotte K Billington and Ian P Hall. Novel cAMP signalling paradigms: therapeutic implications for airway disease (pages 401–410)
■ Jessica Tröger, et al. A-kinase anchoring proteins as potential drug targets (pages 420–433)
■ Paul A Insel, et al. cAMP and Epac in the regulation of tissue fibrosis (pages 447–456)
Fibrosis, the result of excess deposition of extracellular matrix (ECM), in particular collagen, leads to scarring and loss of function in tissues that include the heart, lung, kidney and liver. The second messenger cAMP can inhibit the formation and extent of ECM during this late phase of inflammation, but the mechanisms for these actions of cAMP and of agents that elevate tissue cAMP levels are not well understood. In this article, we review the fibrotic process and focus on two recently recognized aspects of actions of cAMP and its effector Epac (Exchange protein activated by cAMP): (a) blunting of epithelial–mesenchymal transformation (EMT) and (b) down-regulation of Epac expression by profibrotic agents (e.g. TGF-β, angiotensin II), which may promote tissue fibrosis by decreasing Epac-mediated antifibrotic actions. Pharmacological approaches that raise cAMP or blunt the decrease in Epac expression by profibrotic agents may thus be strategies to block or perhaps reverse tissue fibrosis.
■ Kristoffer Watten Brudvik and Kjetil Taskén. Modulation of T cell immune functions by the prostaglandin E2 – cAMP pathway in chronic inflammatory states (pages 411–419)
Cyclic AMP is the intracellular second messenger for a variety of immunoregulatory inflammatory mediators such as prostaglandin E2, adenosine and histamine that signal to effector T cells from monocytes, macrophages and regulatory T cells. Protein kinase A (PKA) type I localizes to lipid rafts in effector T cells during T cell activation and directly modulates proximal signal events including phosphorylation of C-terminal Src kinase (Csk), which initiates a negative signal pathway that fine-tunes the T cell activation process. The PKA-Csk immunoregulatory pathway is scaffolded by the A kinase anchoring protein ezrin, the Csk binding protein phosphoprotein associated with glycosphingolipid-enriched membrane microdomains and the linker protein ezrin/radixin/moesin binding protein of 50 kDa. This pathway is hyperactivated in chronic infections with an inflammatory component such as HIV, other immunodeficiencies and around solid tumours as a consequence of local inflammation leading to inhibition of anti-tumour immunity.
■ Euan Parnell, et al. Regulation of the inflammatory response of vascular endothelial cells by EPAC1 (pages 434–446)
Life-threatening diseases of the cardiovascular system, like atherosclerosis, are exacerbated by unwanted inflammation within the structures of large blood vessels. This inflammation involves increased permeability of the vascular endothelial cells (VECs) that form the lining of blood vessels, leading to exaggerated extravasation of blood components and accumulation of fluid in the extravascular space. This results in tissue dysfunction and increased secretion of chemokines that attract leukocytes and monocytes to the inflamed endothelium. Cyclic AMP is synthesized in VECs in response to endogenous Gs-coupled receptors and is known to limit cytokine action and reduce endothelial hyperpermeability induced by multiple pro-inflammatory stimuli. The mechanisms underlying this anti-inflammatory action of cyclic AMP are now being elucidated and it is becoming clear that the cyclic AMP sensor, exchange protein activated by cyclic AMP (EPAC1), appears to play a key role in suppressing unwanted inflammation. EPAC1 mediates at least three anti-inflammatory pathways in VECs by down-regulating inflammatory signalling through the induction of the suppressors of cytokine signalling 3 (SOCS-3) gene, limiting integrin-dependent vascular permeability and enhancing endothelial barrier function through the stabilization of VE-cadherin junctions. Given that manipulation of cellular cyclic AMP levels currently forms the basis of many effective pharmaceuticals and that EPAC1 is involved in multiple anti-inflammatory protective processes in VECs, does this make EPAC1 an attractive target for the development of activators capable of eliciting a coordinated programme of ‘protection’ against the development of endothelial dysfunction? Here we discuss whether EPAC1 represents an attractive therapeutic target for limiting endothelial dysfunction associated with cardiovascular diseases like atherosclerosis.


Original Article
■ β-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCδ/p38 MAPK signalling in neonatal mouse cardiac fibroblasts (pages 676–688)
心筋線維芽細胞にはアドレナリン作動性β-受容体が存在し,ドパミンやドブタミンによるβ-受容体刺激はEPAC-1を介してPKCδを活性化させ,線維芽細胞の増殖刺激となるばかりか,IL-6などのサイトカインを産生する。
BACKGROUND AND PURPOSE IL-6 plays crucial roles in cardiac hypertrophy, cardiac fibrosis and heart failure. Activation of β-adrenoceptors induced IL-6 production in neonatal mouse cardiac fibroblasts (NMCFs) through a Gs/adenylate cyclase/cAMP/p38 MAPK pathway but independent of PKA. However, how cAMP activates p38 MAPK is still not defined. In this study, we have assessed the role of the exchange protein directly activated by cAMP (Epac) and PKCδ in p38 MAPK activation and IL-6 production by stimulated by the β-adrenoceptor agonist isoprenaline in NMCFs.
EXPERIMENTAL APPROACH The IL-6 concentration in cell culture supernatants was measured by ELISA. The levels of phosphorylated and total p38 MAPK and PKCδ were determined by Western blot analysis. The translocation of PKCδ was determined by immunoblotting the soluble and particulate fractions. Expression of Epac1 or PKCδ was knocked down by the corresponding, adenovirus-mediated, small hairpin RNA (shRNA).
RESULTS In NMCFs, activation of β-adrenoceptors enhanced PKCδ phosphorylation and translocation. Furthermore, knock-down of the PKCδ isoform using an adenovirus-mediated shRNA markedly down-regulated IL-6 induction by NMCFs stimulated with isoprenaline. Moreover, knock-down of Epac1 confirmed that Epac1 was upstream of PKCδ in IL-6 production. Additionally, both Epac1 and PKCδ mediated the p38 MAPK activation induced by isoprenaline.
CONCLUSIONS AND IMPLICATIONS β-Adrenoceptor agonists activate a cAMP/Epac/PKCδ/p38 MAPK pathway to produce IL-6 in NMCFs. This study identifies Epac as the link between cAMP and p38 MAPK signalling pathways and demonstrates that PKCδ can function as a novel downstream effector of this β-adrenoceptor/cAMP/Epac pathway.
■ Michaela Kuhn. Endothelial actions of atrial and B-type natriuretic peptides (pages 522–531)
The cardiac hormone atrial natriuretic peptide (ANP) is critically involved in the maintenance of arterial blood pressure and intravascular volume homeostasis. Its cGMP-producing GC-A receptor is densely expressed in the microvascular endothelium of the lung and systemic circulation, but the functional relevance is controversial. Some studies reported that ANP stimulates endothelial cell permeability, whereas others described that the peptide attenuates endothelial barrier dysfunction provoked by inflammatory agents such as thrombin or histamine. Many studies in vitro addressed the effects of ANP on endothelial proliferation and migration. Again, both pro- and anti-angiogenic properties were described. To unravel the role of the endothelial actions of ANP in vivo, we inactivated the murine GC-A gene selectively in endothelial cells by homologous loxP/Cre-mediated recombination. Our studies in these mice indicate that ANP, via endothelial GC-A, increases endothelial albumin permeability in the microcirculation of the skin and skeletal muscle. This effect is critically involved in the endocrine hypovolaemic, hypotensive actions of the cardiac hormone. On the other hand the homologous GC-A-activating B-type NP (BNP), which is produced by cardiac myocytes and many other cell types in response to stressors such as hypoxia, possibly exerts more paracrine than endocrine actions. For instance, within the ischaemic skeletal muscle BNP released from activated satellite cells can improve the regeneration of neighbouring endothelia. This review will focus on recent advancements in our understanding of endothelial NP/GC-A signalling in the pulmonary versus systemic circulation. It will discuss possible mechanisms accounting for the discrepant observations made for the endothelial actions of this hormone-receptor system and distinguish between (patho)physiological and pharmacological actions. Lastly it will emphasize the potential therapeutical implications derived from the actions of NPs on endothelial permeability and regeneration.
■ EH Kaufman and DB Jacoby. Upping the antedrug: is a novel anti-inflammatory Toll-like receptor 7 agonist also a bronchodilator? (pages 569–572)
In this issue of British Journal of Pharmacology, Biffen and colleagues present a novel Toll-like receptor 7 (TLR7) antedrug to treat allergic disease that is rapidly metabolized in the lung to limit side effects due to systemic exposure. Asthma is characterized as an allergic disease of the lung, and TLR7 agonists are proposed to ameliorate allergic inflammation in the lung, a characteristic of prophylactic medications. We have previously shown that TLR7 agonists of multiple structural classes are acute bronchodilators, characteristic of rescue medication for asthma attacks. It will be interesting to determine whether the bronchodilating effect extends to the novel class of TLR7 agonists described here for a prophylactic and rescue therapy in one drug. Combined with the antedrug approach, this would further limit side effects improving on current combination therapies.
■ M Biffen, et al. Biological characterization of a novel class of toll-like receptor 7 agonists designed to have reduced systemic activity (pages 573–586)
BACKGROUND AND PURPOSE Toll-like receptor 7 (TLR7) agonists have potential in the treatment of allergic diseases. However, the therapeutic utility of current low molecular weight TLR7 agonists is limited by their systemic activity, resulting in unwanted side effects. We have developed a series of TLR7-selective ‘antedrugs’, including SM-324405 and AZ12441970, which contain an ester group rapidly cleaved in plasma to reduce systemic exposure.
EXPERIMENTAL APPROACH Agonist activity at TLR7 of the parent ester and acid metabolite was assessed in vitro in reporter cells and primary cells from a number of species. Pharmacokinetics following a dose to the lungs was assessed in mice and efficacy evaluated in vivo with a mouse allergic airway model.
KEY RESULTS Compounds were selective agonists for TLR7 with no crossover to TLR8 and were metabolically unstable in plasma with the acid metabolite showing substantially reduced activity in a number of assays. The compounds inhibited IL-5 production and induced IFN-α, which mediated the inhibition of IL-5. When dosed into the lung the compounds were rapidly metabolized and short-term exposure of the ‘antedrug’ was sufficient to activate the IFN pathway. AZ12441970 showed efficacy in a mouse allergic airway model with minimal induction of systemic IFN-α, consistent with the low plasma levels of compound.
CONCLUSIONS AND IMPLICATIONS The biological and metabolic profiles of these TLR7-selective agonist ‘antedrug’ compounds are consistent with a new class of compound that could be administered locally for the treatment of allergic diseases, while reducing the risk of systemic side effects.
■ Yimin Yuan, et al. Ethyl pyruvate promotes spinal cord repair by ameliorating the glial microenvironment (pages 749–763)
BACKGROUND AND PURPOSE Spinal cord injury (SCI) triggers a series of endogenous processes, including neuroinflammation and reactive astrogliosis, which may contribute to the failure of neural regeneration and functional recovery. In the present study, the effect of ethyl pyruvate on spinal cord repair was explored.
EXPERIMENTAL APPROACH Functional assessment and histological analyses of astrogliosis, neuroinflammation, neuronal survival and axonal regeneration were performed to investigate the effects of ethyl pyruvate (0.086, 0.215, 0.431 or 0.646 mmol·kg-1·day-1) on spinal cord repair in a rat model of SCI. The effect of ethyl pyruvate (5, 10 or 15 mM) on astrocytic activation was also evaluated in an in vitro‘scratch-wound’ model.
KEY RESULTS Functional assessment showed evident improvement of behavioural functions in the ethyl pyruvate-treated rats. Reactive astrogliosis was significantly inhibited in vivo, after injection of ethyl pyruvate (0.431 mmol·kg-1day-1), and in vitro‘scratch-wound’ model in the presence of 10 or 15 mM ethyl pyruvate. The difference between effective concentration in vitro and in vivo suggests that the inhibitory effect of ethyl pyruvate on astrogliosis in damaged spinal cord is indirect. In addition, ethyl pyruvate (0.431 mmol·kg-1day-1) attenuated SCI-induced neuroinflammation; it decreased the Iba-1-, ED-1- and CD11b-positive cells at the lesion site. Importantly, histological analyses showed a significantly greater number of surviving neurons and regenerative axons in the ethyl pyruvate-treated rats.
CONCLUSIONS AND IMPLICATIONS Ethyl pyruvate was shown to inhibit astrogliosis and neuroinflammation, promote neuron survival and neural regeneration, and improve the functional recovery of spinal cord, indicating a potential neuroprotective effect of ethyl pyruvate against SCI.
■ K Kawamoto, et al. Inhibitory effects of dopamine on spinal synaptic transmission via dopamine D1-like receptors in neonatal rats (pages 788–800)
BACKGROUND AND PURPOSE Dopamine released from the endings of descending dopaminergic nerve fibres in the spinal cord may be involved in modulating functions such as locomotion and nociception. Here, we examined the effects of dopamine on spinal synaptic transmissions in rats.
EXPERIMENTAL APPROACH Spinal reflex potentials, monosynaptic reflex potential (MSR) and slow ventral root potential (sVRP), were measured in the isolated spinal cord of the neonatal rat. Dopamine release was measured by HPLC.
KEY RESULTS Dopamine at lower concentrations (<1 µM) depressed sVRP, which is a C fibre-evoked polysynaptic response and believed to reflect nociceptive transmission. At higher concentrations (>1 µM), in addition to a potent sVRP depression, dopamine depolarized baseline potential and slightly depressed MSR. Depression of sVRP by dopamine was partially reversed by dopamine D1-like but not by D2-like receptor antagonists. SKF83959 and SKF81297, D1-like receptor agonists, and methamphetamine, an endogenous dopamine releaser, also caused the inhibition of sVRP. Methamphetamine also depressed MSR, which was inhibited by ketanserin, a 5-HT2A/2C receptor antagonist. Methamphetamine induced the release of dopamine and 5-HT from spinal cords, indicating that the release of endogenous dopamine and 5-HT depresses sVRP and MSR respectively.
CONCLUSION AND IMPLICATIONS These results suggested that dopamine at lower concentrations preferentially inhibited sVRP, which is mediated via dopamine D1-like and other unidentified receptors. The dopamine-evoked depression is involved in modulating the spinal functions by the descending dopaminergic pathways.
4月よりランチタイム前に1時間~1時間半基礎研究を雑誌1冊報告して頂き,
私が補則解説をし,最終的なレビューをしています。
これを2年間続けると,かなり基本ができます。
2012年3月号のBritish Journal of Pharmacologyを足立裕史先生に担当して頂きましたが,
この号を読むだけで,Gタンパク共役型受容体についてかなり詳細に整理できます。
Gタンパク共役型受容体: molecular pharmacology of GPCRs
総説 Molecular Pharmacology of GPCRs

GPCR expression in tissues and cells: Are the optimal receptors being used as drug targets?
PA Insel, A Snead, F Murray, L Zhang, H Yokouchi, T Katakia, O Kwon, D Dimucci and A Wilderman
British Journal of Pharmacology
Special Issue: Themed Section: Molecular Pharmacology of GPCRs. Guest Editor: Roger J Summers
Volume 165, Issue 6, pages 1613–1616, March 2012
ケモカイン受容体についても非常によくまとめられています
British Journal of Pharmacology 2012;165:1617-1643
DJ Scholten, M Canals, D Maussang, L Roumen, MJ Smit, M Wijtmans, C de Graaf, HF Vischer and R Leurs
Pharmacological modulation of chemokine receptor function
オピオイドの急性耐性についてもまとめられています
British Journal of Pharmacology 2012;165:1704-1716
Vu C Dang and MacDonald J Christie
Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons
clathrin coated vesicle-mediated endocytosisなど
Gタンパク共役型受容体の細胞膜移動トラフィッキングに関与するタンパクが楽しくまとめられています
British Journal of Pharmacology 2012;165:1717-1736
Ana C Magalhaes, Henry Dunn and Stephen SG Ferguson
Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins





