









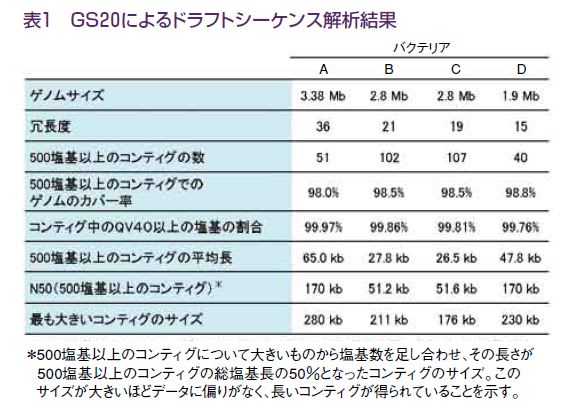
【シーケンスとアノテーション】
シーケンスとアノテーション、翻訳すれば「遺伝子情報の解析とその注釈」となりが丸っぽ、
『デジタル革命渦論』に嵌ってしまう。近年、メタゲノム解析(この言葉も難解だが、難し
い環境中の微生物を培養・増殖させ分析を遺伝子解析で。これらの構造(塩基配列)を網羅的
に調べ、個々の核酸や遺伝子がどの微生物由来かはわからなくとも、環境中の微生物の集合
体がもつ遺伝子群から解明する手法→培養・増殖レス手法)の進展もあって、これまで解析
対象とされてきた微生物種が、実は微生物全体の1%にも満たないことがわかってきた。こ
の膨大な微生物の理解に、これまでに得られているゲノム情報では不十分。さらに数多くの
遺伝子解析が進行中だ。一方、GS20をはじめとする次世代シーケンサーの登場など、シーケ
ンスの技術向上もゲノム解析の増加を後押し。従来の解析法(サンガー法)ではコスト的に
難しかった、変異株・派生株との全ゲノムシーケンスでの比較解析なども現実のものになっ
てきているが、やはり難解だ。ここは情報を整頓。明日の百名山踏破目標の立山チャレンジ
前夜でもあり、次世代シーケンサーと微生物ゲノムアノテーション」の現状をバード・アイ
(雷鳥の)することに。
※ 参考:Next generation sequencer data analysis for microbial genome annotation、大山彰(Oh-
yama,Akira)インシリコバイオロジー㈱(in silico biology,inc.)代表取締役 2006年奈良先端科
学技術犬学院大学情報科学研究科博士課程修士 博士(理学)専門:バイ才サイエンスとイ
ンダストリ- vol.71 No.4 2013)


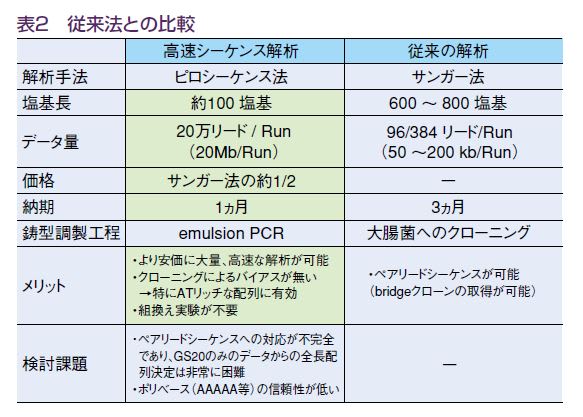
次世代シーケンサー(Next Generation Sequencer: NGS)の急速な普及により微生物ゲノムに
由来する塩基配列が手軽に大量に得られるようになり、微生物株に間するゲノム解析を行
う機会が増えると予想されている。従来、NGS の出力データからゲノム解析を実行するに
は解析阻害・障壁が存在。処理時間の長さ、処理に必要な大きなメモリーサイズ、使用す
る多くのソフトウェアがLinux環境のみで動作するなどがの障壁があった。このため、これ
らの処理は情報解析担当者に依頼していたが、ボーダレスとなり、比較的容易にNGSデータ
ゲノム解析を遂行できるようになった。NGSから直接出力されるリード塩基配列は通常遺
伝子より短い断片配列であり、そのままでは遺伝子解析に利用できない。このため、短い
リード塩基配列同士を互いの相同性を利用して結合し、由来するゲノム遺伝子の全塩基配
列を再構成する。これを de novo アセンブリ(以後アセンブル)と呼んでいる。アセンブ
リによって遺伝子長より大きなゲノムDNA塩基配列が得られると、この膨大なゲノム塩基
配列上のどこにどのような生物学的特徴があるかの同定/処理をゲノムアノテーションと呼
んでいる。

NGSから直接出力されるリード塩基配列は通常遺伝子より短い断片配列であり、そのまま
では遺伝子解析に利用できない。このため、短いリード塩基配列同士を互いの相同性を利用
して結合し、由来するゲノムDNAの全塩基配列を再構成する。これを de novoアセンブリ(=
アセンブル)と呼ぶ。アセンブリによって遺伝子長より大きなゲノムDNA塩基配列が得られ
ると、この膨大なゲノム塩基配列上のどこにどのような生物学的特徴があるかを同定する。
この処理をゲノムアノテーションと呼ぶ。各注釈は、解析をより効率的に進めるには、ゲノ
ム地図上の様々な種類の道標としてグラフィカルに表示できることが求められている(第4
次産業=図画像形成産業)。この機能がゲノムビューアだ。ゲノム地図上に新たな実験やデ
ータ解析からの編集機能が必要となる。この編集操作機能をゲノム地図エディタだ。NSG
出力データは、新規ゲノム配列解析→変異株のシーケンシング→多数の変異断片を参照ゲノ
ム塩基配列上にラベリング→塩基レベルでのゲノム変異解析が可能となる。また、異なる生
育環境における対象徹生物のRNAサンプルをNGSでシーケンシング、マッピングで、遺
伝子発現解析も可能となりこれをマッピングと呼んでいる。

ところで、アセンブリにより長いゲノム塩基配列が得られると、その塩基配列上に生物学的
な特徴(Feature)を同定し、注釈として記録するゲノムアノテーションを行うが、一般に同定
される生物学的な特徴は遺伝子配列。これらの生物学的特徴をゲノム塩基配列から同定する
遺伝子予測ソフトウェアもLinux上で動作が多いが、ORF同定機能だけに絞っても多種類のソ
フトウェアあるが、原核微生物ゲノムには、MetaGeneAnnotator、酵母やカビなどの真核微生
物ゲノムにはAUGUSTUSを使用。原核徹生物の遺伝子同定精度高く、約90%以上の精度を有
すが、真核徹生物の遺伝手同定精度が高くない。特に適当な既知近縁種ゲノムの学習用デー
タが存在しない真核微生物の de novo遺伝子同定精度はかなり低い。この場合タンパク質由
来のアミノ酸配列を使用し、アミノ酸配列とそのゲノム配列との相同性から遺伝子を同定す
る方法(アミノ酸マッピングと呼ぶ)の併用で同定精度が向上する。tRNAの間定には、99%以
上の予測精度を持つと言われるtRNAScan-SEが広く使用されている。rRNA同定には、de novo
同定法の1つとしてRNAmmerが使用される場合が多い。

既知のrRNAとの相同性を検出する方法でもrRNAの範囲は決定できるが、rRNA末端を正確
に予測するには、正確なrRNAデータベースの構築が必要で、原核微生物ゲノム上には1Mbp
当たり1,000個程度のORFが同定される。同定されたORFそれぞれについて、データベースに
登録されている全生物のORFのアミノ酸配列全体を参照データとして使用して相同性解析を
実行。データベースにヒットした配列に記述された注釈を転記し、新規ゲノム上の遺伝子に
この注釈のコピーが記録され全自動アノテーションが実行するが、この自動アノテーション
結果は完全ではない(この自動アノテーション結果を「ドラフトアノテーション」と呼ぶ)。
登録済ORFのアミノ酸配列数も急速に増大。相同性解析にかかる時間も比例して増大。この
程度の相同性解析でも小さなPC上で実行可能であるが、結果を得るには長時間を必要とする。
これらの処理時間を短縮したい場合、大型クラスター計算機上に実装された微生物ゲノムア
ノテーションサービスは直ちにドラフトアノテーションできる。MiGAP(Microbial Genome An-
notation Pipeline)は国立遺伝学研究所の大規模スパコン上に実装された高速並列化微生物ゲノ
ムアノテーションサービスである。MiGAPを使用すると、E.coli クラスの微生物ゲノム1個
を数時間で全自動アノテーションできる。MiGAPパイプラインヘの役人データは、アセンブ
リで生成された全コンティグ配列(マルチプルFastAフォーマット)で、これを直接役人できる。
現在は、同時に4つのゲノムを並列処理することが可能で、最大数千個のORFの相同性解析
が同時に処理されている。5個目以降の投入ゲノムは実行中のゲノムアノテーションが終了
するまで処理待ち行列に入れ、相同性解析に使用される注釈付配列データベースは7種類(C
OGなどのオーソログデータベース3種とRefSeq Protein、NRAA、およびTrEMBLである.また
これとは別に生物種別にGenBankを使用できる。そのうち3種を選択して、3段階の相同性
検索を自動実行できる。処理時間をさらに短縮するため、前段階で注釈がついた場合は後段
階での相同性解析をスキップできる分岐機能も実装する。また参照データベースの一部は定
期的に最新のリリースに替わるため、その前後でアノテーション結果が異なる可能性がある。
これを避けるために、MiGAPでは過去のリリースを特に指定してアノテーションの実行が可
能。 MiGAPサービスは国立遺伝学研究所DDBJにユーザ登録すれば誰でも利用できる。ゲノ
ム投入方法は極めて簡単で、操作方法やアノテーションに関する予備知識なしに自動ドラフ
トアノテーション結果を得られるという。
さらに、自動アノテーション処理にはLinux 環境が必要であるが、Linux計算機への遺伝子同
定ソフトウェアインストールおよび日常の保守にはLinuxの操作知識が必要で、実験研究者に
は敷居が高かったが、研究者が日常使用しているWindowsPCやMac上に仮想的なLinux OS を
インストールすることが可能であり、これを利用して個人用のアセンブリ・アノテーション
処理用Linux環境を実現。Linuxでのみ動作するアノテーション用ソフトウェアを操作するには、
Linuxのコマンドライン操作を知る必要があった。WindowsやMacで動作するゲノム解析ソフ
トウェアであるIMO(In sikico MolecularCloning)およびGT(Genome Traveler)上に、これらの
Linuxコマンドを使用せずに簡単なグラフィカルな操作で仮想的Linux OS上にインストール
された遺伝子同定ソフトウェア群を実行できる機能を実装した。これにより、MiGAPサービ
スから得られる結果とぼけ同等のアノテーションを自分のPC上で自動実行できるようになる。
NGSでゲノムシーケンシングし、それらのリード塩基配列を基準株ゲノム塩基配列上にマ
ッピングすることで、ゲノム変異解析ができる一方で、ゲノムシーケンシングに加えてRNA
シーケンシング(RNA-Seq)すれば、リード配列をそのゲノム塩基配列上にマッピングするこ
とにより、de novo 遺伝子予測に頼らず、高精度にエクソンィントロン境界を同定すること
が可能な遺伝子同定かでき、遺伝子の発現解析が可能だ。マッピング結果は通常、使用した
参照ゲノム配列に対して貼り付けたリード塩基配列をアラィンメント(並置)して表示するこ
とのできる(リード)アラィンメントビューアが必要である。以下にGTにおけるリードアラ
ィンメント、マッピングを通常のPC上で実行するには処理時間がかかる。GTにはSlideSortと
いう独自のソフトウェアを実装されており、これを用いて通常のPC上で短時間にNGSショ
ートリードがマッピングできる。


この後、 「ゲノムビューア・エディタ、ゲノム解析および詳細アノテーション」でドラフ
トアノテーション配列をゲノムビューア・エディタに読み込み、そのゲノム地図を閲覧しな
がら多様な解析を行い、解析結果を注釈に反映することができ、多数の近縁種ゲノムをNG
Sシーケンシングすることにより、ゲノム間の比較解析ができるようになる。これを比較ゲノム解析と
いう。 ドラフトに手を加え詳細なアノテーションを行うのに役に立つ比較ゲノム解析手法など俯瞰す
る必要があるが、ここで時間切れ、残件はまたの機会にチャレンジすることに。





