








以下はバイオインフォマティックス初心者のウエット系研究者がかいた覚書である。
まず準備として
ネットブックで行うRNA-seqデーター解析(3)でLT-HSCとST-HSCのデータがcuffdiffで処理されて、LT_HSC_VS_ST_HSC_cuffdiff_resultというフォルダにはいっているはずである。
またネットブックで行うRNA-seqデーター解析(3.5)でその可視化ソフトであるcummeRbundがRにインストールされたはずである。
今回はこのcummeRbundを用いて可視化を試みることとする(*)
もしこれまでに触ったことがなければ、RおよびcummeRbundのコマンドの概要を少し知っておいた方がよい。
Rに関しては二階堂愛さんのRの基礎が少し難しいがよいだろう(なお練習問題は必ずしも解けなくても大丈夫。オブジェクト指向の概要とコマンドの概要がわかればよい)。
cummeRbundに関してはinsilicodb.orgが英語だが割と実用的でよい。日本語のものはcummeRbund manualをそのまま訳したものが多く今一つ使いにくいが、
牧場の朝さんのと
二階堂愛さんのRNA-seqデーター解析
がわりかし使いよい。
まずRを起動しcummeRbundを立ち上げてみる
>library(cummeRbund)
次にLT_HSC_VS_ST_HSC_cuffdiff_resultフォルダから、diffという拡張子がついたデーターファイルをcuffというオブジェクトによみこむ(**)。
> cuff <- readCufflinks("LT_HSC_VS_ST_HSC_cuffdiff_result")
うまく読み込めていれば
>cuff
とやるとその概要を次のように表示してくれるはずである。
CuffSet instance with:
4 samples
23306 genes
30563 isoforms
25977 TSS
22856 CDS
139494 promoters
155862 splicing
113886 relCDS
遺伝子(genes)、アイソフォーム(isoforms)、転写開始点(TSS)などなどの情報。
ここでちょっとした分析を行うなら、
>csBoxplot(genes(cuff))
とやると各群のboxplotを表示してくれる(***)

>csDensity(genes(cuff))
とやると、densityplotを表示してくれる

さらに
> csDendro(genes(cuff))
とやると、樹形図を表示してくれる

そして
>csVolcanoMatrix(genes(cuff))
とやると、ボルケーノプロットを表示してくれる

次にgenediffというオブジェクトに差のある遺伝子データ(genes)を読み込む。
> genediff <- diffData(genes(cuff))
全部を出すとちょっとめんどうなので、その最初の部分だけを表示させると
>head(genediff)
gene_id sample_1 sample_2 status value_1 value_2 log2_fold_change
1 0610005C13Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
2 0610007N19Rik q1 q2 OK 0.0000 3.78706 1.79769e+308
3 0610007P14Rik q1 q2 OK 197.9410 83.90660 -1.23821e+00
4 0610008F07Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
5 0610009B14Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
6 0610009B22Rik q1 q2 OK 94.5476 105.75900 1.61668e-01
test_stat p_value q_value significant
1 0.00000e+00 1.0000000 1.000000 no
2 1.79769e+308 0.1660150 0.533321 no
3 1.79123e+00 0.0732564 0.492534 no
4 0.00000e+00 1.0000000 1.000000 no
5 0.00000e+00 1.0000000 1.000000 no
6 -1.81075e-01 0.8563090 0.968486 no
このようなデーター構成になっていることがわかる。一番前にgene_idがあることに注目してこの差のある遺伝子のIDを取ってくると、
>genediffdataID <- genediff_data[,1]
として、
>genediffdataID
(前略)
[2293] "Tmem180" "Tmem194" "Tmem38a" "Traf3ip2"
[2297] "Trdmt1" "Trim23" "Tstd3" "Ttll1"
[2301] "Ubald1" "Ube2e2" "Ubxn11" "Vpreb3"
[2305] "Wdr12" "Wnk1" "Xist" "Zbtb34"
[2309] "Zfp354a" "Zfp446" "Zfp523" "Zfp551"
[2313] "Zfp605" "Zfp810" "Zfp846" "Zfp90"
[2317] "Zfp92" "Zfyve19" "Zscan2"
とちゃんとIDがとれていることがわかる。
このgenediffdataIDは列ベクトル(Columun Vector)であるが、このIDをもとにデーターを取ってくる作業をするためには行ベクトル(Row Vector)である必要があり、転置変換を行う(****)。
> genediffdataID <- t(genediffdataID)
そしてmyGenesというオブジェクトにcuffというオブジェクトからこのgenediffdataIDを持つ遺伝子の情報だけとってくると、
>myGenes <-getGenes(cuff, genediffdataID)
Getting gene information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting isoforms information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting CDS information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting TSS information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting promoter information:
distData
Getting splicing information:
distData
Getting relCDS information:
distData
とデーターがとってこられる。
例えばmyGenesをもとにヒートマップを書かせてみると、
> csHeatmap(myGenes)
遺伝子が多すぎてわかりませんが、次のようになる。

myGenesをもとに4群でクラスター分析すると、
>k.means <-csCluster(myGenes, k=4)
その状況をグラフに書かせてみると、
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot

q1,q2:LT-HSC
q3,q4:ST-HSC
であったので、もしあなたが血液学者なら、もっとも未分化な血液幹細胞LT-HSCで多く出ている遺伝子が何かしりたいところだろう。このクラスターのうち、q1,q2での発現が,q3,q4より多い、クラスター2のデーターがほしくなるはずである。
>k.means
とやると、クラスター分析のデーターをだしてくれ
(前略)
Mpst 2 3 0.4569913751
Ino80b 2 3 0.4508379672
Zfp697 2 3 0.4463841894
5730577I03Rik 2 3 0.4462468051
Ttc23 2 1 0.4423819910
Bai3 2 1 0.4386712043
Sh3pxd2a 2 3 0.4293128230
Nab2 2 3 0.4288460352
Jag1 2 4 0.4265375878
(中略)
C77080 2 4 -0.2102395161
D5Ertd605e 2 4 -0.2228394545
Srr 2 4 -0.2498411870
(後略)
とどんな遺伝子かわかる。
この遺伝子群のヒートマップをたとえば書かせてみるには、この遺伝子群のIDをエクセルにコピペして(*****)、タブ区切りのtxtファイルをつくる。
それをRに読み込んで、転置し
> CL2ID <- read.table("CL2.txt" , header=F, sep="t")
> CL2ID <- t(CL2ID)
データーをcuffから、Genes_CL2にとってきて、
>Genes_CL2 <-getGenes(cuff, CL2ID)
>csHeatmap(Genes_CL2)

やっぱ数が多いなあー。
そこで発現量が高く(status列がOK)、統計的に優位なものだけ(significant列がyes)のものだけgenediffオブジェクトからとってくることとする(******)。
> genediff_data <- genediff[((genediff$status == 'OK')& (genediff$significant == 'yes')),]
なおこの操作をおこなうとgenediffオブジェクトのデーターは一部失われる。(*******)
IDをとって、データーをmyGenes2にいれ
> genediffdataID2 <- genediff_data[,1]
> genediffdataID2 <- t(genediffdataID2)
> myGenes2 <-getGenes(cuff, genediffdataID2)
4群のクラスター分析を行い
> k.means <-csCluster(myGenes2, k=4)
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot
図をかかせてみると、
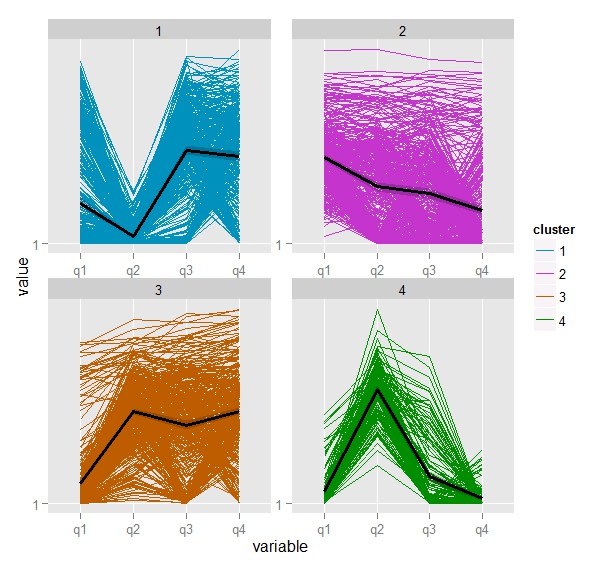
やはり遺伝子数はまだ多そうである。
統計的にいいのかどうかはわからないが6群でクラスター分析を行い
プロットすると、
> k.means <-csCluster(myGenes2, k=6)
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot

クラスター6が面白そうである。
>k.means
でこの遺伝子がなにかしらべ、やや多いので上から50個をエクセルでCL6-50.txtファイルにいれ
> CL6ID <- read.table("CL6-50.txt" , header=F, sep="t")
> CL6ID <- t(CL6ID)
> Genes_CL6 <-getGenes(cuff, CL6ID)
> csHeatmap(Genes_CL6)
とやると

わりとみられる感じになる(********)。
また
>csHeatmap(Genes_CL6, cluster ='both')
とやると遺伝子の側でもクラスタリングしてくれる。

ふーっ!なんだかなー。
(*)ネットブックで行うRNA-seqデーター解析(4)としたかったのだが、ちょっとまだまとめきれていないので、IT編とした。
(**)この操作がRの基本である。
(***)cuffdiffでラベルがうまくつけられなかったつけがここにでている。q1,q2,q3,q4...
あと一部の群で0の値があり、それが少しデーターをおかしくしている気がする。
(****)そのままやると変なエラーメッセージがでます。
(*****)このあたりがド素人的な所以。。
(******)insilicodb.orgにあるスクリプトのようにp値(α値)で区切ってもよいのかもしれない。
ちなみにα=0.05をカットオフにして、ヒートマップを書かせると
> mySigGeneIds<-getSig(cuff,alpha=0.05,level='genes')
> myGenes3<-getGenes(cuff,mySigGeneIds)
> csHeatmap(myGenes3,cluster='both')
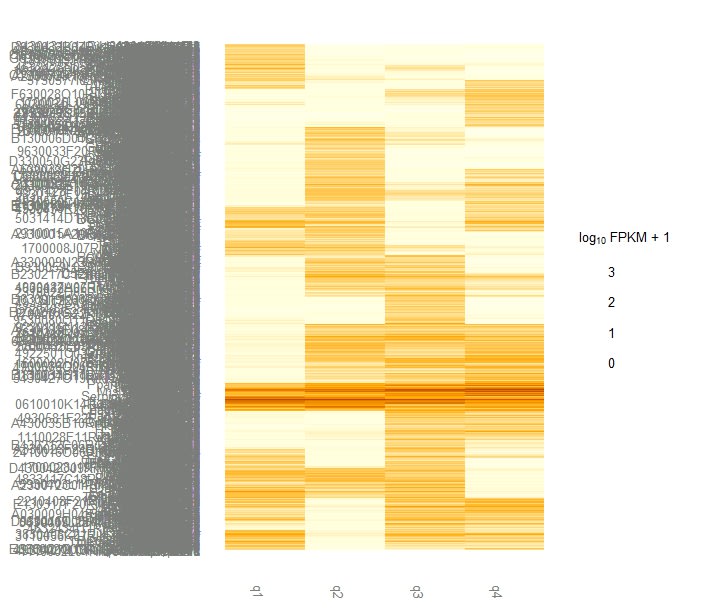
のようになる。まだまだやのうー!
(*******)条件にあったものが消えている気がする。
(********)gene_idが2重に表示される意味が分からない。
まず準備として
ネットブックで行うRNA-seqデーター解析(3)でLT-HSCとST-HSCのデータがcuffdiffで処理されて、LT_HSC_VS_ST_HSC_cuffdiff_resultというフォルダにはいっているはずである。
またネットブックで行うRNA-seqデーター解析(3.5)でその可視化ソフトであるcummeRbundがRにインストールされたはずである。
今回はこのcummeRbundを用いて可視化を試みることとする(*)
もしこれまでに触ったことがなければ、RおよびcummeRbundのコマンドの概要を少し知っておいた方がよい。
Rに関しては二階堂愛さんのRの基礎が少し難しいがよいだろう(なお練習問題は必ずしも解けなくても大丈夫。オブジェクト指向の概要とコマンドの概要がわかればよい)。
cummeRbundに関してはinsilicodb.orgが英語だが割と実用的でよい。日本語のものはcummeRbund manualをそのまま訳したものが多く今一つ使いにくいが、
牧場の朝さんのと
二階堂愛さんのRNA-seqデーター解析
がわりかし使いよい。
まずRを起動しcummeRbundを立ち上げてみる
>library(cummeRbund)
次にLT_HSC_VS_ST_HSC_cuffdiff_resultフォルダから、diffという拡張子がついたデーターファイルをcuffというオブジェクトによみこむ(**)。
> cuff <- readCufflinks("LT_HSC_VS_ST_HSC_cuffdiff_result")
うまく読み込めていれば
>cuff
とやるとその概要を次のように表示してくれるはずである。
CuffSet instance with:
4 samples
23306 genes
30563 isoforms
25977 TSS
22856 CDS
139494 promoters
155862 splicing
113886 relCDS
遺伝子(genes)、アイソフォーム(isoforms)、転写開始点(TSS)などなどの情報。
ここでちょっとした分析を行うなら、
>csBoxplot(genes(cuff))
とやると各群のboxplotを表示してくれる(***)
>csDensity(genes(cuff))
とやると、densityplotを表示してくれる
さらに
> csDendro(genes(cuff))
とやると、樹形図を表示してくれる
そして
>csVolcanoMatrix(genes(cuff))
とやると、ボルケーノプロットを表示してくれる
次にgenediffというオブジェクトに差のある遺伝子データ(genes)を読み込む。
> genediff <- diffData(genes(cuff))
全部を出すとちょっとめんどうなので、その最初の部分だけを表示させると
>head(genediff)
gene_id sample_1 sample_2 status value_1 value_2 log2_fold_change
1 0610005C13Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
2 0610007N19Rik q1 q2 OK 0.0000 3.78706 1.79769e+308
3 0610007P14Rik q1 q2 OK 197.9410 83.90660 -1.23821e+00
4 0610008F07Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
5 0610009B14Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
6 0610009B22Rik q1 q2 OK 94.5476 105.75900 1.61668e-01
test_stat p_value q_value significant
1 0.00000e+00 1.0000000 1.000000 no
2 1.79769e+308 0.1660150 0.533321 no
3 1.79123e+00 0.0732564 0.492534 no
4 0.00000e+00 1.0000000 1.000000 no
5 0.00000e+00 1.0000000 1.000000 no
6 -1.81075e-01 0.8563090 0.968486 no
このようなデーター構成になっていることがわかる。一番前にgene_idがあることに注目してこの差のある遺伝子のIDを取ってくると、
>genediffdataID <- genediff_data[,1]
として、
>genediffdataID
(前略)
[2293] "Tmem180" "Tmem194" "Tmem38a" "Traf3ip2"
[2297] "Trdmt1" "Trim23" "Tstd3" "Ttll1"
[2301] "Ubald1" "Ube2e2" "Ubxn11" "Vpreb3"
[2305] "Wdr12" "Wnk1" "Xist" "Zbtb34"
[2309] "Zfp354a" "Zfp446" "Zfp523" "Zfp551"
[2313] "Zfp605" "Zfp810" "Zfp846" "Zfp90"
[2317] "Zfp92" "Zfyve19" "Zscan2"
とちゃんとIDがとれていることがわかる。
このgenediffdataIDは列ベクトル(Columun Vector)であるが、このIDをもとにデーターを取ってくる作業をするためには行ベクトル(Row Vector)である必要があり、転置変換を行う(****)。
> genediffdataID <- t(genediffdataID)
そしてmyGenesというオブジェクトにcuffというオブジェクトからこのgenediffdataIDを持つ遺伝子の情報だけとってくると、
>myGenes <-getGenes(cuff, genediffdataID)
Getting gene information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting isoforms information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting CDS information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting TSS information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting promoter information:
distData
Getting splicing information:
distData
Getting relCDS information:
distData
とデーターがとってこられる。
例えばmyGenesをもとにヒートマップを書かせてみると、
> csHeatmap(myGenes)
遺伝子が多すぎてわかりませんが、次のようになる。
myGenesをもとに4群でクラスター分析すると、
>k.means <-csCluster(myGenes, k=4)
その状況をグラフに書かせてみると、
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot
q1,q2:LT-HSC
q3,q4:ST-HSC
であったので、もしあなたが血液学者なら、もっとも未分化な血液幹細胞LT-HSCで多く出ている遺伝子が何かしりたいところだろう。このクラスターのうち、q1,q2での発現が,q3,q4より多い、クラスター2のデーターがほしくなるはずである。
>k.means
とやると、クラスター分析のデーターをだしてくれ
(前略)
Mpst 2 3 0.4569913751
Ino80b 2 3 0.4508379672
Zfp697 2 3 0.4463841894
5730577I03Rik 2 3 0.4462468051
Ttc23 2 1 0.4423819910
Bai3 2 1 0.4386712043
Sh3pxd2a 2 3 0.4293128230
Nab2 2 3 0.4288460352
Jag1 2 4 0.4265375878
(中略)
C77080 2 4 -0.2102395161
D5Ertd605e 2 4 -0.2228394545
Srr 2 4 -0.2498411870
(後略)
とどんな遺伝子かわかる。
この遺伝子群のヒートマップをたとえば書かせてみるには、この遺伝子群のIDをエクセルにコピペして(*****)、タブ区切りのtxtファイルをつくる。
それをRに読み込んで、転置し
> CL2ID <- read.table("CL2.txt" , header=F, sep="t")
> CL2ID <- t(CL2ID)
データーをcuffから、Genes_CL2にとってきて、
>Genes_CL2 <-getGenes(cuff, CL2ID)
>csHeatmap(Genes_CL2)
やっぱ数が多いなあー。
そこで発現量が高く(status列がOK)、統計的に優位なものだけ(significant列がyes)のものだけgenediffオブジェクトからとってくることとする(******)。
> genediff_data <- genediff[((genediff$status == 'OK')& (genediff$significant == 'yes')),]
なおこの操作をおこなうとgenediffオブジェクトのデーターは一部失われる。(*******)
IDをとって、データーをmyGenes2にいれ
> genediffdataID2 <- genediff_data[,1]
> genediffdataID2 <- t(genediffdataID2)
> myGenes2 <-getGenes(cuff, genediffdataID2)
4群のクラスター分析を行い
> k.means <-csCluster(myGenes2, k=4)
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot
図をかかせてみると、
やはり遺伝子数はまだ多そうである。
統計的にいいのかどうかはわからないが6群でクラスター分析を行い
プロットすると、
> k.means <-csCluster(myGenes2, k=6)
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot
クラスター6が面白そうである。
>k.means
でこの遺伝子がなにかしらべ、やや多いので上から50個をエクセルでCL6-50.txtファイルにいれ
> CL6ID <- read.table("CL6-50.txt" , header=F, sep="t")
> CL6ID <- t(CL6ID)
> Genes_CL6 <-getGenes(cuff, CL6ID)
> csHeatmap(Genes_CL6)
とやると
わりとみられる感じになる(********)。
また
>csHeatmap(Genes_CL6, cluster ='both')
とやると遺伝子の側でもクラスタリングしてくれる。
ふーっ!なんだかなー。
(*)ネットブックで行うRNA-seqデーター解析(4)としたかったのだが、ちょっとまだまとめきれていないので、IT編とした。
(**)この操作がRの基本である。
(***)cuffdiffでラベルがうまくつけられなかったつけがここにでている。q1,q2,q3,q4...
あと一部の群で0の値があり、それが少しデーターをおかしくしている気がする。
(****)そのままやると変なエラーメッセージがでます。
(*****)このあたりがド素人的な所以。。
(******)insilicodb.orgにあるスクリプトのようにp値(α値)で区切ってもよいのかもしれない。
ちなみにα=0.05をカットオフにして、ヒートマップを書かせると
> mySigGeneIds<-getSig(cuff,alpha=0.05,level='genes')
> myGenes3<-getGenes(cuff,mySigGeneIds)
> csHeatmap(myGenes3,cluster='both')
のようになる。まだまだやのうー!
(*******)条件にあったものが消えている気がする。
(********)gene_idが2重に表示される意味が分からない。






Thanks for sharing,
Alain
Thank you for your comment.
I also appreciate the information in InSilicoDB.org.
It is really helpful for everyone who is involved in RNAseq analysis.
I will check your new system later.