同様の内容をIT編で書いた。ブラッシュアップしたいのだが、現時点でまだできていないので、現状を載せておく(スペース確保のため)。
ネットブックで行うRNA-seqデーター解析(3)でLT-HSCとST-HSCのデータがcuffdiffで処理されて、LT_HSC_VS_ST_HSC_cuffdiff_resultというフォルダにはいっているはずである。
また
ネットブックで行うRNA-seqデーター解析(3.5)でその可視化ソフトであるcummeRbundがRにインストールされたはずである。
今回はこのcummeRbundを用いて可視化を試みることとする(*)
なお準備としてRおよびcummeRbundのコマンドの概要を少し知っておいた方がよい。
Rに関しては
二階堂愛さんのRの基礎が少し難しいがよいだろう(なお練習問題は必ずしも解けなくても大丈夫。オブジェクト指向の概要とコマンドの概要がわかればよい)。
cummeRbundに関しては
insilicodb.orgが英語だが割と実用的でよい。日本語のものは
cummeRbund manualをそのまま訳したものが多く今一つ使いにくいが、
牧場の朝さんのと
二階堂愛さんのRNA-seqデーター解析
がわりかし使いよい。
まずLT_HSC_VS_ST_HSC_cuffdiff_resultフォルダから、diffという拡張子がついたデーターファイルをcuffというオブジェクトによみこむ(**)。
> cuff <- readCufflinks("LT_HSC_VS_ST_HSC_cuffdiff_result")
うまく読み込めていれば
>cuff
とやるとその概要を次のように表示してくれるはずである。
CuffSet instance with:
4 samples
23306 genes
30563 isoforms
25977 TSS
22856 CDS
139494 promoters
155862 splicing
113886 relCDS
遺伝子(genes)、アイソフォーム(isoforms)、転写開始点(TSS)などなどの情報。
ここでちょっとした分析を行うなら、
>csBoxplot(genes(cuff))
とやると各群のboxplotを表示してくれる(***)

>csDensity(genes(cuff))
とやると、densityplotを表示してくれる

さらに
> csDendro(genes(cuff))
とやると、樹形図を表示してくれる

そして
>csVolcanoMatrix(genes(cuff))
とやると、ボルケーノプロットを表示してくれる

次にgenediffというオブジェクトに差のある遺伝子データ(genes)を読み込む。
> genediff <- diffData(genes(cuff))
全部を出すとちょっとめんどうなので、その最初の部分だけを表示させると
>head(genediff)
gene_id sample_1 sample_2 status value_1 value_2 log2_fold_change
1 0610005C13Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
2 0610007N19Rik q1 q2 OK 0.0000 3.78706 1.79769e+308
3 0610007P14Rik q1 q2 OK 197.9410 83.90660 -1.23821e+00
4 0610008F07Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
5 0610009B14Rik q1 q2 NOTEST 0.0000 0.00000 0.00000e+00
6 0610009B22Rik q1 q2 OK 94.5476 105.75900 1.61668e-01
test_stat p_value q_value significant
1 0.00000e+00 1.0000000 1.000000 no
2 1.79769e+308 0.1660150 0.533321 no
3 1.79123e+00 0.0732564 0.492534 no
4 0.00000e+00 1.0000000 1.000000 no
5 0.00000e+00 1.0000000 1.000000 no
6 -1.81075e-01 0.8563090 0.968486 no
このようなデーター構成になっていることがわかる。一番前にgene_idがあることに注目してこの差のある遺伝子のIDを取ってくると、
>genediffdataID <- genediff_data[,1]
として、
>genediffdataID
(前略)
[2293] "Tmem180" "Tmem194" "Tmem38a" "Traf3ip2"
[2297] "Trdmt1" "Trim23" "Tstd3" "Ttll1"
[2301] "Ubald1" "Ube2e2" "Ubxn11" "Vpreb3"
[2305] "Wdr12" "Wnk1" "Xist" "Zbtb34"
[2309] "Zfp354a" "Zfp446" "Zfp523" "Zfp551"
[2313] "Zfp605" "Zfp810" "Zfp846" "Zfp90"
[2317] "Zfp92" "Zfyve19" "Zscan2"
とちゃんとIDがとれていることがわかる。
このgenediffdataIDは列ベクトル(Columun Vector)であるが、このIDをもとにデーターを取ってくる作業をするためには行ベクトル(Row Vector)である必要があり、転置変換を行う(****)。
> genediffdataID <- t(genediffdataID)
そしてmyGenesというオブジェクトにcuffというオブジェクトからこのgenediffdataIDを持つ遺伝子の情報だけとってくると、
>myGenes <-getGenes(cuff, genediffdataID)
Getting gene information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting isoforms information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting CDS information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting TSS information:
FPKM
Differential Expression Data
Annotation Data
Replicate FPKMs
Counts
Getting promoter information:
distData
Getting splicing information:
distData
Getting relCDS information:
distData
とデーターがとってこられる。
例えばmyGenesをもとにヒートマップを書かせてみると、
> csHeatmap(myGenes)
遺伝子が多すぎてわかりませんが、次のようになる。

myGenesをもとに4群でクラスター分析すると、
>k.means <-csCluster(myGenes, k=4)
その状況をグラフに書かせてみると、
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot

q1,q2:LT-HSC
q3,q4:ST-HSC
であったので、もしあなたが血液学者なら、もっとも未分化な血液幹細胞LT-HSCで多く出ている遺伝子が何かしりたいところだろう。このクラスターのうち、q1,q2での発現が,q3,q4より多い、クラスター2のデーターがほしくなるはずである。
>k.means
とやると、クラスター分析のデーターをだしてくれ
(前略)
Mpst 2 3 0.4569913751
Ino80b 2 3 0.4508379672
Zfp697 2 3 0.4463841894
5730577I03Rik 2 3 0.4462468051
Ttc23 2 1 0.4423819910
Bai3 2 1 0.4386712043
Sh3pxd2a 2 3 0.4293128230
Nab2 2 3 0.4288460352
Jag1 2 4 0.4265375878
(中略)
C77080 2 4 -0.2102395161
D5Ertd605e 2 4 -0.2228394545
Srr 2 4 -0.2498411870
(後略)
とどんな遺伝子かわかる。
この遺伝子群のヒートマップをたとえば書かせてみるには、この遺伝子群のIDをエクセルにコピペして(*****)、タブ区切りのtxtファイルをつくる。
それをRに読み込んで、転置し
> CL2ID <- read.table("CL2.txt" , header=F, sep="t")
> CL2ID <- t(CL2ID)
データーをcuffから、Genes_CL2にとってきて、
>Genes_CL2 <-getGenes(cuff, CL2ID)
>csHeatmap(Genes_CL2)

これだと数が多いのでちょっとヒートマップで何かを言うことが難しい。
そこで発現量が高く(status列がOK)、統計的に優位なものだけ(significant列がyes)のものだけgenediffオブジェクトからとってくることとする(******)。
> genediff_data <- genediff[((genediff$status == 'OK')& (genediff$significant == 'yes')),]
なおこの操作をおこなうとgenediffオブジェクトのデーターは一部失われる。(*******)
IDをとって、データーをmyGenes2にいれ
> genediffdataID2 <- genediff_data[,1]
> genediffdataID2 <- t(genediffdataID2)
> myGenes2 <-getGenes(cuff, genediffdataID2)
4群のクラスター分析を行い
> k.means <-csCluster(myGenes2, k=4)
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot
図をかかせてみると、
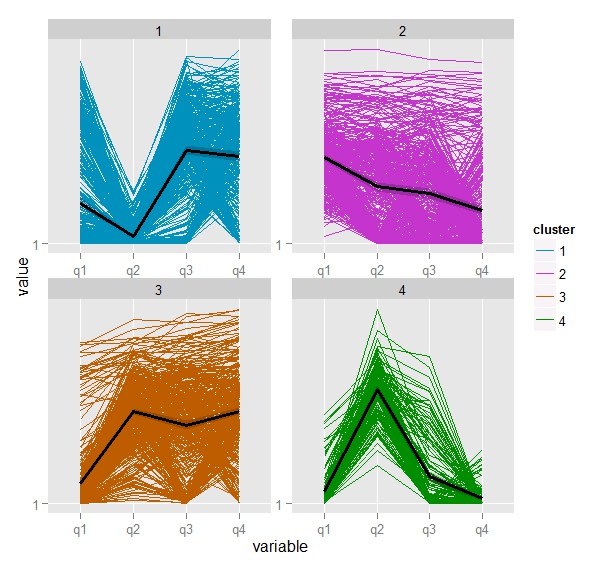
やはり遺伝子数はまだ多そうである。
統計的にいいのかどうかはわからないが6群でクラスター分析を行い
プロットすると、
> k.means <-csCluster(myGenes2, k=6)
> k.means.plot <- csClusterPlot(k.means)
> k.means.plot

クラスター6が面白そうである。
>k.means
でこの遺伝子がなにかしらべ、やや多いので上から50個をエクセルでCL6-50.txtファイルにいれ
> CL6ID <- read.table("CL6-50.txt" , header=F, sep="t")
> CL6ID <- t(CL6ID)
> Genes_CL6 <-getGenes(cuff, CL6ID)
> csHeatmap(Genes_CL6)
とやると

わりとみられる感じになる(********)。
また
>csHeatmap(Genes_CL6, cluster ='both')
とやると遺伝子の側でもクラスタリングしてくれる。

もう少しブラッシュアップしないといけないであろうが何とか、LT-HSCに高そうな遺伝子が取れてきているようである。ただ本当にValidなものなのかは、ほかのアレーやRNA-seqデーターを参照して分析しないといけないであろう。。
(*)ネットブックで行うRNA-seqデーター解析(4)としたかったのだが、ちょっとまだまとめきれていないので、IT編とした。
(**)この操作がRの基本である。
(***)cuffdiffでラベルがうまくつけられなかったつけがここにでている。q1,q2,q3,q4...
あと一部の群で0の値があり、それが少しデーターをおかしくしている気がする。
(****)そのままやると変なエラーメッセージがでます。
(*****)このあたりがド素人的な所以。。
(******)
insilicodb.orgにあるスクリプトのようにp値(α値)で区切ってもよいのかもしれない。
ちなみにα=0.05をカットオフにして、ヒートマップを書かせると
> mySigGeneIds<-getSig(cuff,alpha=0.05,level='genes')
> myGenes3<-getGenes(cuff,mySigGeneIds)
> csHeatmap(myGenes3,cluster='both')
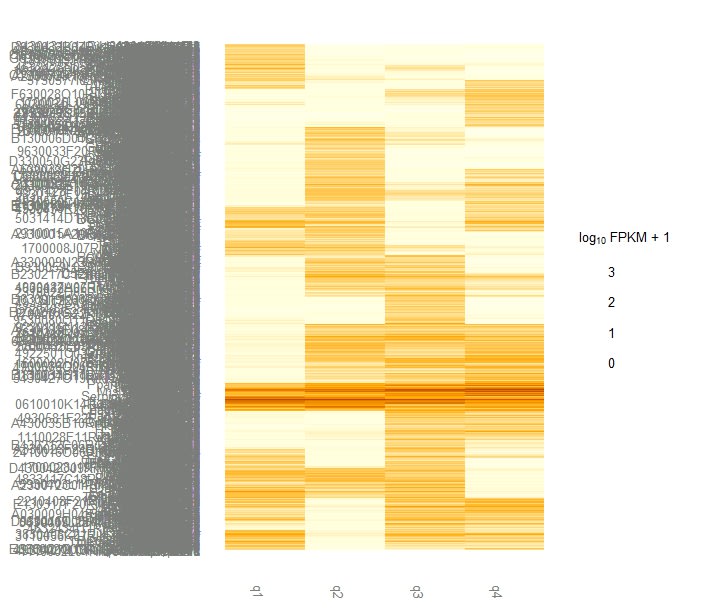
のようになる。まだまだやのうー!
(*******)条件にあったものが消えている。
(********)gene_idが2重に表示される理由がまだ分からない。








