門田先生の著書「トランスクリプトーム解析」が素晴らしい
先日東大の門田幸二先生の「トランスクリトーム解析」を手に入れた。全くの初心者にはちょっと難易度が高い本であるが、少し慣れてきた人にとっては使い勝手の良い本である。
アレーデーターの読み込み&正規化
先日のhuman白血病幹細胞がうまくクラスタリングされないのではないか?という問題をもう少しエクステンシブに扱ってみた。前回の解析ではすでにプロセスされているデーターテーブルを使ったが、まずアレーファイルを全部読み込んで自分で正規化なども行ってみることとする。
まず読み込みであるが、RでArrayExpressというライブラリーをつかってつぎのように読み込むことができる。
>#installing ArrayExpress
>source("https://bioconductor.org/biocLite.R")
>biocLite("ArrayExpress")
>#library(ArrayExpress)
>#get raw data stored in GSE30375
>param <- "GSE30375"
>hoge <- getAE(param,type="raw",extract=F)
これでワーキングディレクトリにE-GEOD-30375.raw.1.zipができているはずである。これを解凍してやると、ワーキングデレクトリに幾つかのCELファイルができる。解凍はRでもできるようなのだが、いまひとつうまくいかないので、Macのterminalで
> mkdir GSE30375
> unzip E-GEOD-30375.raw.1.zip
> mv *.CEL GSE30375
としてGSE30375というフォルダにうつす。
その後自分的はワーキングディレクトリを、GSE30375に移して(ここからR)
> setwd
>#normalization of the raw files with MAS5 and write a table for the out put
>out_f <- "hoge1.txt"
>hoge <- ReadAffy()
>eset <- mas5(hoge)
でMAS5による正規化が終わる。
発現情報は
>data <- exprs(eset)
で取り出せる。
ファイルに落としたければ
>out_f <- GSE30375_MAS.txt
>write.exprs(eset,file=out_f)
でよい。
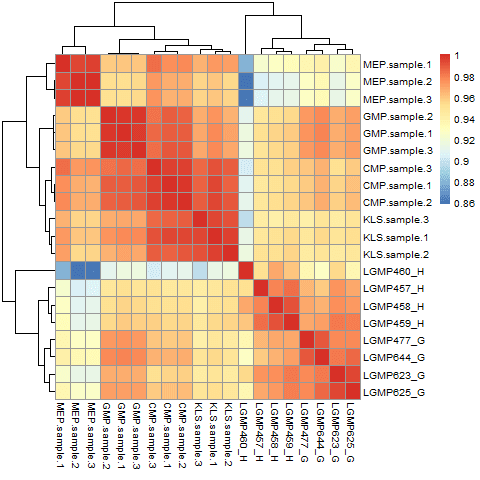
pheatmapによる解析
前にもやったようにpheatmapで解析すると、
>library(pheatmap)
>y <- cor(data)
>pheatmap(y)
これだとちょっとみにくいので
>pheatmap(y,show_colnames=F,fontsize=6)

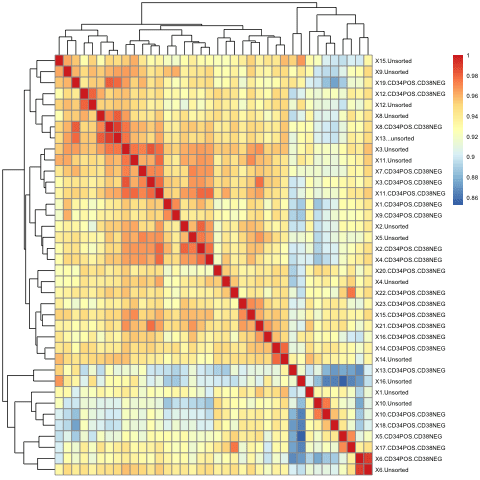
やっぱり正規化をあたらてめて行っても、うまく白血病幹細胞CD34+CD38-がわかれていなさそうである!
そこで少しサンプル数をしぼってみると
サンプル数15

サンプル数10

サンプル数6

サンプル数3

といった形で、同じ番号(たとえばX1)のサンプル(同じ患者由来)がクラスタリングしやすく、白血病幹細胞かどうかといったことはあまりクラスタリングに影響しないようであった。
ちなみにCD34+CD38-とunsortedのAMLサンプルだけを比較しても同じようなことが言える。
hclustによる解析
門田先生の本に習って、その他のクラスタリングもためしてみる。
1-spearmanだけやってみましたが
>data.dist <- as.dist(1-cor(data,method= "spearman"))
>out <- hclust(data.dist, method = "average")
>plot(out)

のようにCD34+CD38-とunsortedのAMLサンプルでクラスタリングさせても、2群うまく分かれてくれない。門田先生の記述によると、このような場合には2つの群の遺伝子発現があまり違わない時に起こるようである!白血病幹細胞はバルクの白血病細胞と比べて違わなくてもよいのかと思う。。
ちなみにここからはきちんと理解していないが、differentiallyに発現する遺伝子(DEG)を抽出してくると、
>source("https://bioconductor.org/biocLite.R")
>biocLite("limma")
>data <- log2(data)
>colnames(data) <- c(paste("LSC_",1;23,sep=""),paste("Bulk_",1:15,sep=""))
>data.c1 <- c(rep(1,23),rep(2,15))
>design <- model.matrix(~ as.factor(data.c1))
>fit <- lmFit(data,design)
>out <- eBayes(fit)
>p.value <- out$p.value[,ncol(design)]
>q.value <- p.adjust(p.value,method="BH")
>ranking <- rank(p.value)
>tmp <- cbind(rownames(data),data,p.value,q.value,ranking)
>write.table(tmp,file="AML_MAS_DEG.txt",sep="\t",append=F,quote=F,row.names=F)
>topTable(out,coef=colnames(design)[ncol(design)],adjust="BH",number=8)

となって優位にp<0.01かつFDR<0.1を満たすDEGは2個しかなく、クラスタリングからも想定されたように、少なくともこのデーターセットに関しては、白血病幹細胞の特異的な遺伝子発現というものは観察できないようである。
先日東大の門田幸二先生の「トランスクリトーム解析」を手に入れた。全くの初心者にはちょっと難易度が高い本であるが、少し慣れてきた人にとっては使い勝手の良い本である。
アレーデーターの読み込み&正規化
先日のhuman白血病幹細胞がうまくクラスタリングされないのではないか?という問題をもう少しエクステンシブに扱ってみた。前回の解析ではすでにプロセスされているデーターテーブルを使ったが、まずアレーファイルを全部読み込んで自分で正規化なども行ってみることとする。
まず読み込みであるが、RでArrayExpressというライブラリーをつかってつぎのように読み込むことができる。
>#installing ArrayExpress
>source("https://bioconductor.org/biocLite.R")
>biocLite("ArrayExpress")
>#library(ArrayExpress)
>#get raw data stored in GSE30375
>param <- "GSE30375"
>hoge <- getAE(param,type="raw",extract=F)
これでワーキングディレクトリにE-GEOD-30375.raw.1.zipができているはずである。これを解凍してやると、ワーキングデレクトリに幾つかのCELファイルができる。解凍はRでもできるようなのだが、いまひとつうまくいかないので、Macのterminalで
> mkdir GSE30375
> unzip E-GEOD-30375.raw.1.zip
> mv *.CEL GSE30375
としてGSE30375というフォルダにうつす。
その後自分的はワーキングディレクトリを、GSE30375に移して(ここからR)
> setwd
>#normalization of the raw files with MAS5 and write a table for the out put
>out_f <- "hoge1.txt"
>hoge <- ReadAffy()
>eset <- mas5(hoge)
でMAS5による正規化が終わる。
発現情報は
>data <- exprs(eset)
で取り出せる。
ファイルに落としたければ
>out_f <- GSE30375_MAS.txt
>write.exprs(eset,file=out_f)
でよい。
pheatmapによる解析
前にもやったようにpheatmapで解析すると、
>library(pheatmap)
>y <- cor(data)
>pheatmap(y)
これだとちょっとみにくいので
>pheatmap(y,show_colnames=F,fontsize=6)
やっぱり正規化をあたらてめて行っても、うまく白血病幹細胞CD34+CD38-がわかれていなさそうである!
そこで少しサンプル数をしぼってみると
サンプル数15
サンプル数10
サンプル数6
サンプル数3
といった形で、同じ番号(たとえばX1)のサンプル(同じ患者由来)がクラスタリングしやすく、白血病幹細胞かどうかといったことはあまりクラスタリングに影響しないようであった。
ちなみにCD34+CD38-とunsortedのAMLサンプルだけを比較しても同じようなことが言える。
hclustによる解析
門田先生の本に習って、その他のクラスタリングもためしてみる。
1-spearmanだけやってみましたが
>data.dist <- as.dist(1-cor(data,method= "spearman"))
>out <- hclust(data.dist, method = "average")
>plot(out)
のようにCD34+CD38-とunsortedのAMLサンプルでクラスタリングさせても、2群うまく分かれてくれない。門田先生の記述によると、このような場合には2つの群の遺伝子発現があまり違わない時に起こるようである!白血病幹細胞はバルクの白血病細胞と比べて違わなくてもよいのかと思う。。
ちなみにここからはきちんと理解していないが、differentiallyに発現する遺伝子(DEG)を抽出してくると、
>source("https://bioconductor.org/biocLite.R")
>biocLite("limma")
>data <- log2(data)
>colnames(data) <- c(paste("LSC_",1;23,sep=""),paste("Bulk_",1:15,sep=""))
>data.c1 <- c(rep(1,23),rep(2,15))
>design <- model.matrix(~ as.factor(data.c1))
>fit <- lmFit(data,design)
>out <- eBayes(fit)
>p.value <- out$p.value[,ncol(design)]
>q.value <- p.adjust(p.value,method="BH")
>ranking <- rank(p.value)
>tmp <- cbind(rownames(data),data,p.value,q.value,ranking)
>write.table(tmp,file="AML_MAS_DEG.txt",sep="\t",append=F,quote=F,row.names=F)
>topTable(out,coef=colnames(design)[ncol(design)],adjust="BH",number=8)
となって優位にp<0.01かつFDR<0.1を満たすDEGは2個しかなく、クラスタリングからも想定されたように、少なくともこのデーターセットに関しては、白血病幹細胞の特異的な遺伝子発現というものは観察できないようである。