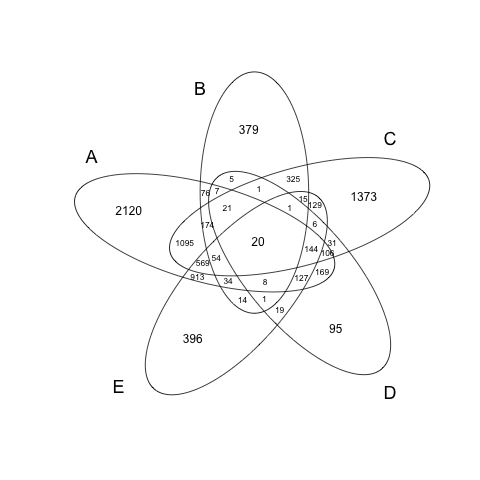
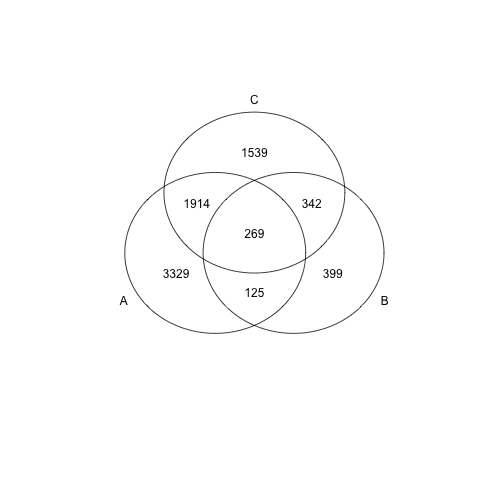
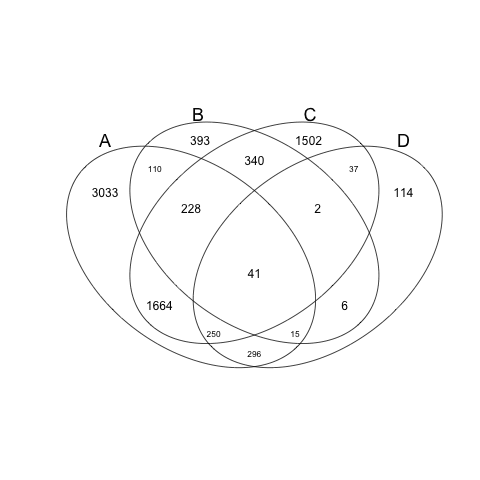
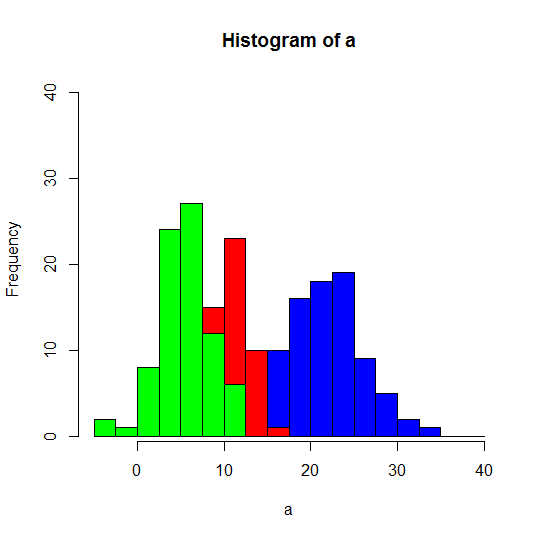
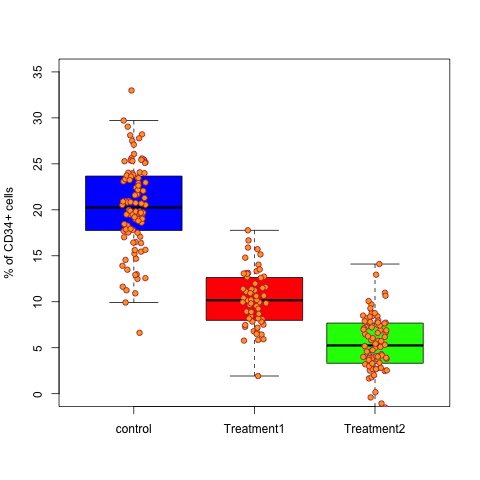
Rでheatmapを作るのに,gplotsパッケージのheatmap.2を使うのがつかいよい。
一番わかりやすいのが、Mannheimiaさんのブログである。
基本的には
> install.packages("gplots")
>library(gplots)
で読み込んで、お好みのdataについてheatmapを書かせればよい。
>heatmap.2(as.matrix(data), col=greenred(75), scale="row", key=T, keysize=1.5,
density.info="none", trace="none",cexCol=0.9, cexRow=0.5)
個人的にはメモリのカラーはgreenredでRowも提示したほうがいいので、以上のようなコマンドが気に入っている。
あと入れるデーターについては、彼の説明がうまいのでいかに引用しておく。
また追記で補足します!
However this is not the way of dealing with the data. When we work with high throughput data, the first step is to log-transform the intensities and then apply a normalization method. We can do that with the following lines:
data = log2(data)
boxplot(data, cex.axis=0.8, las=2, main="Original distribution of data",
ylab="Log2(Intensity)") # Draw a boxplot.
# Normalization
library(preprocessCore)
data2 = normalize.quantiles(as.matrix(data)) # A quantile normalization.
# Copy the row and column names from data to data2:
rownames(data2) = rownames(data)
colnames(data2) = colnames(data)
boxplot(data2, cex.axis=0.8, las=2, main="Distribution after normalization",
ylab="Log2(Intensity)")
# t-test using the limma package:
library(limma)
design = cbind(Cell_A = c(1,1,1,0,0,0,0,0,0), # First 3 columns->Cell_A
Cell_B = c(0,0,0,1,1,1,0,0,0),
Cell_C = c(0,0,0,0,0,0,1,1,1)) # Last 3 columns->Cell_C
fit = lmFit(data2, design=design) # Fit the original matrix to the above design.
# We want to compare A vs. B, A vs. C and B vs. C
contrastsMatrix = makeContrasts("Cell_A-Cell_B","Cell_A-Cell_C","Cell_B-Cell_C",
levels = design)
fit2 = contrasts.fit(fit, contrasts = contrastsMatrix) # Making the comparisons.
fit2 = eBayes(fit2) # Moderating the t-tetst by eBayes method.
一番わかりやすいのが、Mannheimiaさんのブログである。
基本的には
> install.packages("gplots")
>library(gplots)
で読み込んで、お好みのdataについてheatmapを書かせればよい。
>heatmap.2(as.matrix(data), col=greenred(75), scale="row", key=T, keysize=1.5,
density.info="none", trace="none",cexCol=0.9, cexRow=0.5)
個人的にはメモリのカラーはgreenredでRowも提示したほうがいいので、以上のようなコマンドが気に入っている。
あと入れるデーターについては、彼の説明がうまいのでいかに引用しておく。
また追記で補足します!
However this is not the way of dealing with the data. When we work with high throughput data, the first step is to log-transform the intensities and then apply a normalization method. We can do that with the following lines:
data = log2(data)
boxplot(data, cex.axis=0.8, las=2, main="Original distribution of data",
ylab="Log2(Intensity)") # Draw a boxplot.
# Normalization
library(preprocessCore)
data2 = normalize.quantiles(as.matrix(data)) # A quantile normalization.
# Copy the row and column names from data to data2:
rownames(data2) = rownames(data)
colnames(data2) = colnames(data)
boxplot(data2, cex.axis=0.8, las=2, main="Distribution after normalization",
ylab="Log2(Intensity)")
# t-test using the limma package:
library(limma)
design = cbind(Cell_A = c(1,1,1,0,0,0,0,0,0), # First 3 columns->Cell_A
Cell_B = c(0,0,0,1,1,1,0,0,0),
Cell_C = c(0,0,0,0,0,0,1,1,1)) # Last 3 columns->Cell_C
fit = lmFit(data2, design=design) # Fit the original matrix to the above design.
# We want to compare A vs. B, A vs. C and B vs. C
contrastsMatrix = makeContrasts("Cell_A-Cell_B","Cell_A-Cell_C","Cell_B-Cell_C",
levels = design)
fit2 = contrasts.fit(fit, contrasts = contrastsMatrix) # Making the comparisons.
fit2 = eBayes(fit2) # Moderating the t-tetst by eBayes method.