Peter Adorjan, Jurgen Distler, Evelyne Lipscher, Fabian Model, Jurgen Muller, Cecile Pelet, Aron Braun, Andrea R. Florl, David Gutig, Gabi Grabs, Andre Howe, Mischo Kursar, Ralf Lesche, Erik Leu, Andre Lewin, Sabine Maier, Volker Muller, Thomas Otto, Christian Scholz, Wolfgang A. Schulz, Hans-Helge Seifert, Ina Schwope, Heike Ziebarth, Kurt Berlin, Christian Piepenbrock and Alexander Olek
Tumour class prediction and discovery by microarray-based DNA methylation analysis
Nucleic Acids Research, 2002, Vol. 30, No. 5 e21
[PDF]
・一般的に広く用いられているmRNAに基づく発現量解析ではなく、DNA methylation of CpG sitesに基づいた解析の紹介。
・データ:2クラス×6組(Female×Male, Healthy T and B cells×T-ALL/B-ALL, T-ALL/B-ALL×AML, BPH×Prostate carcinoma, Healthy kidney×Kidney carcinoma, Prostate×Kidney)、18~38サンプル
・遺伝子ランキング法:Two sample t-test
・Class prediction:SVM
・Class discovery:Hierarchical clustering
・結果「We confirmed the general assumption that massively parallel analysis is in most cases superior to the use of low-dimensional data sets. Nevertheless, in some cases computational selection of informative features out of an initially high-deimensional space allows subsequent classification through a low-dimensional approach.」
・問題点「Major problems, therefore, are the limitation to sites for which methylation-sensitive enzymes are available, the inability to analyse a set of specific candidate genes, the occurence of false positives due to incomplete digestion and the large amount of high molecular weight DNA required. In addition, most of these techniques are highly labour intensive and cannot be automated.」
・問題点「Methylation-specific PCR is highly sensitive but not quantitative, primer design is very labour intensive and false positives occur frequently.」
・従来法「However, in expression profiling signal intensities strongly depend on both the absolute and relative amounts of the different mRNA species and thus comparison between independent mRNA species and thus comarison between independent experiments is difficult.」
・利点「This greatly improves the comparability of the results and therefore enables the screening of larger populations, as is needed for example in multi-centre trials and prospective studies.」
・DNA methylationとは何なのか、生化学的知識がないので歯が立たず。
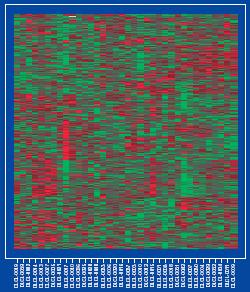
・今更ながら、マイクロアレイのデータで男女の判別が出来ることにちょっとオドロキ(図)。
Tumour class prediction and discovery by microarray-based DNA methylation analysis
Nucleic Acids Research, 2002, Vol. 30, No. 5 e21
[PDF]
・一般的に広く用いられているmRNAに基づく発現量解析ではなく、DNA methylation of CpG sitesに基づいた解析の紹介。
・データ:2クラス×6組(Female×Male, Healthy T and B cells×T-ALL/B-ALL, T-ALL/B-ALL×AML, BPH×Prostate carcinoma, Healthy kidney×Kidney carcinoma, Prostate×Kidney)、18~38サンプル
・遺伝子ランキング法:Two sample t-test
・Class prediction:SVM
・Class discovery:Hierarchical clustering
・結果「We confirmed the general assumption that massively parallel analysis is in most cases superior to the use of low-dimensional data sets. Nevertheless, in some cases computational selection of informative features out of an initially high-deimensional space allows subsequent classification through a low-dimensional approach.」
・問題点「Major problems, therefore, are the limitation to sites for which methylation-sensitive enzymes are available, the inability to analyse a set of specific candidate genes, the occurence of false positives due to incomplete digestion and the large amount of high molecular weight DNA required. In addition, most of these techniques are highly labour intensive and cannot be automated.」
・問題点「Methylation-specific PCR is highly sensitive but not quantitative, primer design is very labour intensive and false positives occur frequently.」
・従来法「However, in expression profiling signal intensities strongly depend on both the absolute and relative amounts of the different mRNA species and thus comparison between independent mRNA species and thus comarison between independent experiments is difficult.」
・利点「This greatly improves the comparability of the results and therefore enables the screening of larger populations, as is needed for example in multi-centre trials and prospective studies.」
・DNA methylationとは何なのか、生化学的知識がないので歯が立たず。
・今更ながら、マイクロアレイのデータで男女の判別が出来ることにちょっとオドロキ(図)。