Mark Smolkin and Debashis Ghosh
Cluster stability scores for microarray data in cancer studies
BMC Bioinformatics 2003, 4:36 doi:10.1186/1471-2105-4-36
[PDF][Web Site]
・サンプルのクラス分けについて、そのクラスの数ではなく、安定度(Cluster stability scores; Random subspace methods)で評価する。
・データ
1.Childhood cancer [Khan]
2.B-Lymphoma [Alizadeh]
3.Cutaneous melanoma [Bittner]
・クラス分け法:Hierarchical clustering (Average linkage, Complete linkage)
・比較したクラス分けの評価法
1.R-index [McShane]
2.The cluster scoring method [Tibshirani]
★実験1:クラス数を既知として処理
★実験2:クラス数を未知として処理
・使用した "R" のコードは以下からダウンロード可能。
http://www.sph.umich.edu/~ghoshd/COMPBIO/CSS/
・問題点「While most work has focused on estimating the number of clusters in a dataset, the question of stability of individual-level clusters has not been addressed.」
・概要「We address this problem by developing cluster stability scores using subsampling techniques. These scores exploit the redundancy in biologically discriminatory information on the chip. Our approach is generic and can be used with any clustering method.」
・手順「Two approaches are taken in this paper. For the first, we assume that the number of clusters is known; sensitivity measures using random subspace methods are calculated. In the second situation, the number of clusters is unknown. We address this problem by proposing a two-stage procedure in which the number of clusters is estimated at the first stage and sensitivity measures are calculated at the second.」
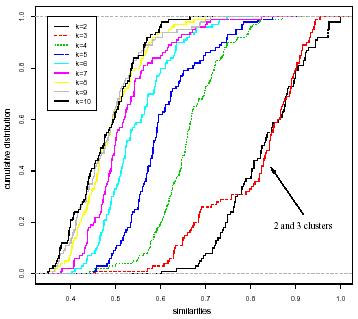
・サンプル全体からランダムに一部のサンプルを抽出しクラス分け。これを繰り返して、同じ結果になる率が高いクラスほど安定度の点数が高くなる、ということらしい(間違ってるかも)。
Cluster stability scores for microarray data in cancer studies
BMC Bioinformatics 2003, 4:36 doi:10.1186/1471-2105-4-36
[PDF][Web Site]
・サンプルのクラス分けについて、そのクラスの数ではなく、安定度(Cluster stability scores; Random subspace methods)で評価する。
・データ
1.Childhood cancer [Khan]
2.B-Lymphoma [Alizadeh]
3.Cutaneous melanoma [Bittner]
・クラス分け法:Hierarchical clustering (Average linkage, Complete linkage)
・比較したクラス分けの評価法
1.R-index [McShane]
2.The cluster scoring method [Tibshirani]
★実験1:クラス数を既知として処理
★実験2:クラス数を未知として処理
・使用した "R" のコードは以下からダウンロード可能。
http://www.sph.umich.edu/~ghoshd/COMPBIO/CSS/
・問題点「While most work has focused on estimating the number of clusters in a dataset, the question of stability of individual-level clusters has not been addressed.」
・概要「We address this problem by developing cluster stability scores using subsampling techniques. These scores exploit the redundancy in biologically discriminatory information on the chip. Our approach is generic and can be used with any clustering method.」
・手順「Two approaches are taken in this paper. For the first, we assume that the number of clusters is known; sensitivity measures using random subspace methods are calculated. In the second situation, the number of clusters is unknown. We address this problem by proposing a two-stage procedure in which the number of clusters is estimated at the first stage and sensitivity measures are calculated at the second.」
・サンプル全体からランダムに一部のサンプルを抽出しクラス分け。これを繰り返して、同じ結果になる率が高いクラスほど安定度の点数が高くなる、ということらしい(間違ってるかも)。