









□ Deep learning for computational biology:
>> http://msb.embopress.org/content/12/7/878
refined hyper‐parameter space can be further explored by random sampling, settings w/ the best performance on the validation set are chosen. Frameworks such as Spearmint, Hyperopt or SMAC allow to automatically explore the hyper‐parameter space using Bayesian optimization. However, although conceptually more powerful, they are at present more difficult to apply and parallelize than random sampling.
□ RSAT matrix-clustering: dynamic exploration & redundancy reduction of transcription factor binding motif collections
>> http://biorxiv.org/content/early/2016/07/27/065565
Meta-databases merging these collections do not offer non-redundant versions, because automatically regrouping similar motifs into clusters cannot be easily achieved with available tools. matrix-clustering is a versatile tool that clusters similar TFBMs into multiple trees, and automatically creates non-redundant collections of motifs.
□ Codon usage is a stochastic process across genetic codes of the kingdoms of life:
>> http://biorxiv.org/content/early/2016/07/27/066381
This stochastic model provides accurate, one-parameter characterizations of 2210 nuclear & mitochondrial genomes represented w/ >10^4 codons. Lopsided usages are also widely distributed across genomes but less frequent. a cross-species, cross-kingdom model provides a universal framework for investigating determinants of codon use.
□ MethFlowVM: a virtual machine for the integral analysis of bisulfite sequencing data:
>> http://biorxiv.org/content/early/2016/07/31/066795
The goal is to allow the user profile the bisulfite sequencing data locally and compare it to all tissues and pathophysiological conditions stored in NGSmethDB without the necessity to upload the user data to any public severs.
□ Modeling translation elongation dynamics by deep learning reveals new insights into landscape of ribosome stalling:
>> http://biorxiv.org/content/biorxiv/early/2016/08/02/067108.full.pdf
ROSE can greatly outperform gkm-SVM with an increase in the area under the receiver operating characteristic curve (AUROC) by up to 18.4%.
a complete CNN in our deep learning framework can be formulated as
p(s) = sigm(concati=1,2,3(pooli(ReLUi(convi(encode(s)))))),
where i represents the kernel index in the parallel architecture, and encode(·), conv(·), ReLU(·), pool(·), concat(·) and sigm(·) represent the one-hot encoding, convolution, ReLU, max pooling, concatenation and sigmoid operations, respectively.
□ Spherical: an iterative workflow for assembling metagenomic datasets:
>> http://biorxiv.org/content/biorxiv/early/2016/08/02/067256.full.pdf
□ ABySS 2.0: Resource-Efficient Assembly of Large Genomes using a Bloom Filter:
>> http://biorxiv.org/content/early/2016/08/07/068338
The main innovation of ABySS 2.0.0 is a Bloom filter-based implementation of the de Bruijn graph assembly algorithm that reduces the overall memory requirements of ABySS by an order of magnitude. The Chromium scaffolding increased the scaffold NG50 of ABySS 2.0.0 assembly from 26.9 Mbp to 41.9 Mbp.
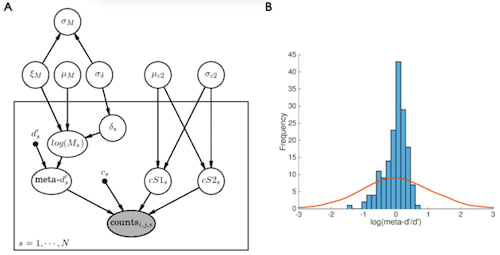
□ Hierarchical Bayesian estimation of metacognitive efficiency from confidence ratings:
>> http://biorxiv.org/content/early/2016/08/09/068601
It naturally incorporates variable uncertainty about finite hit and false-alarm rates; HMeta-d framework to estimate trial-level effects may therefore accelerate understanding of the neural basis of metacognitive efficiency.
□ petal – Co-expression network modelling in R:
>> http://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-016-0298-8
Mathematically, the expression profile of a gene is an n-dimensional vector. Association between each gene pair (two n-dimensional vectors) is computed via an association measure, transforming the m×n expression matrix into an m×m symmetric association matrix. The resulting network model should follow typical properties of complex networks such as scale-free and small-world.
□ DeepWAS: Directly integrating regulatory information into GWAS using deep learning supports master regulator MEF2C:
>> http://biorxiv.org/content/early/2016/08/11/069096
deepWAS approach is superior to simply overlapping peaks or tracks of chromatin features to GWAS signals, even though it is computationally more expensive due to the deep learning-based predictions, cross-validated LASSO models and permutation-based model selection.
□ SEX-DETector: a probabilistic approach to study sex chromosomes in non-model organisms:
>> http://gbe.oxfordjournals.org/content/early/2016/08/03/gbe.evw172.short
□ Highly parallel direct RNA sequencing on an array of nano pores:
>> http://biorxiv.org/content/early/2016/08/12/068809
Recurrent Neural Networks (RNNs) have been shown to be effective at basecalling MinION with DNA and could equally be applied to RNA-seq. the state space of an HMM to include states for modified bases, or to allow an RNN to provide classification scores for a larger kmer set, Although the computational cost of HMMs will grow exponentially with the number of bases included in the model. an array of nanopores to sequence RNA directly, a combination of being amplification-free, capable of detecting nucleotide analogues, and the method is strand-specific, allowing the unambiguous identification of sense and antisense transcripts.
□ Oxford Nanopore Develops Direct RNA Sequencing Protocol:
>> https://www.genomeweb.com/sequencing/oxford-nanopore-develops-direct-rna-sequencing-protocol
□ Moon Shots Program APOLLO Project to Perform DNA/RNA-Seq on Serial Biopsies:
>> https://www.mdanderson.org/newsroom/2016/08/moon-shots-program-a0.html
□ Better Approximation Algorithms for Scaffolding Problems:
>> http://link.springer.com/chapter/10.1007/978-3-319-39817-4_3
wish to find a Hamiltonian path P in G such that all edges of D appear in P and the total weight of edges in P but not in D is maximized. This problem is NP-hard and the previously best polynomial-time approximation algorithm for it achieves a ratio of 1/2. design a new polynomial-time approximation algorithm achieving a ratio of 5−5ϵ/9−8ϵ for any constant 0<ϵ

□ An Automated Microwell Platform for Large-Scale Single Cell RNA-Seq:
>> http://biorxiv.org/content/biorxiv/early/2016/08/18/070193.full.pdf
the compatibility of this system with the simple, 3’-end library preparation scheme SCRB-Seq and the barcoded “Drop-Seq” capture beads.
□ Reinforcement Learning algorithms for regret minimization in structured Markov Decision Processes:
>> http://arxiv.org/pdf/1608.04929v1.pdf
three algorithms, pUCB, pThompson and warmPSRL, that treat ‘structured policies’ as arms of a corresponding multi-arm bandit (MAB) problem. Algorithms that pursue regret minimization in a finite time horizon are the UCRL, PSRL, Thompson PSRL and the RLPA algorithms.
□ Critical Behavior from Deep Dynamics: A Hidden Dimension in Natural Language:
>> https://arxiv.org/pdf/1606.06737v2.pdf
There are close analogies between this deep recursive grammar and more conventional physical systems. hidden depth dimension corresponds to cosmic time, & the parameter which labels the place in the sequence of interest corresponds to space. deep generative grammar model can be viewed as an idealization of a long-short term memory (LSTM) recurrent neural net, where the “forget weights” drop with depth so that the forget timescales grow exponentially with depth.
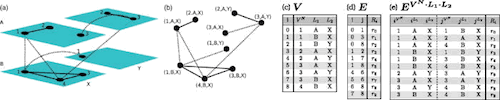
□ Deep graphs: A general framework to analyze heterogeneous complex systems across scales
>> http://scitation.aip.org/content/aip/journal/chaos/26/6/10.1063/1.4952963
Deep graphs provided a natural and straightforward way to identify large clusters of extreme precipitation events, track temporal resolution. Intersection partitions not only allow to derive a tensor-like representation of a multilayer network and also allow to calculate similarity. the auxiliary connector & selector functions enable to create & select super edges, thereby allowing to forge the topology of a deep graph.
□ Supporting R9 data in nano polish:
>> http://simpsonlab.github.io/2016/08/23/R9/
The R9 pore model was the simplest issue to resolve - ONT provided a new table mapping k-mers to Gaussian parameters. the ONT basecaller would now use a recurrent neural network rather than a hidden Markov model. Supporting this data would require quite a few changes to nanopolish - a set of parameters for the Gaussian emission distributions in HMM.
□ Numerical Analysis of the Immersed Boundary Method for Cell-Based Simulation:
>> http://biorxiv.org/content/early/2016/08/24/071423
This approach considers the dynamics of elastic membranes, representing cell boundaries, immersed in a viscous Newtonian fluid. this implementation scales linearly with time step, and subquadratically with mesh spacing and immersed boundary node spacing.
□ Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation
>> http://biorxiv.org/content/early/2016/08/24/071282
Canu is the only tool capable of assembling low-accuracy 1D Nanopore data, while scaling to gigabase-sized genomes. Canu supports Sun Grid Engine, Simple Linux Utility for Resource Management (SLURM), Load Sharing Facility(LSF), Portable Batch System (PBS).
□ Statistical inference for time course RNA-Seq data using a negative binomial mixed-effect model:
>> http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1180-9
the Markov property states that the conditional dependency of prior information from all time, can be simplified to the conditional dependency of prior information of k time points. The p values for identifying NPDE and PDE genes are calculated through a permutation procedure. compute a Kullback-Leibler distance ratio KLR for a gene, the time labels for the gene are shuffled & recompute the statistic for the gene.
a nonparametric mixed-effect model with logit link
log{p(ti,g,k)/(1−p(ti,g,k))}=log(βti,g)+η(ti,g)+zkbk,
exon level read counts of DE genes, were generated using the following smooth function,
η(ti,g)=sin(2.5π((0.9−2ti)I[g=2]+ti)+2.

□ “First DNA sequencing in space.”
>> http://go.nasa.gov/2bV2UnD
□ RGFA: powerful and convenient handling of assembly graphs:
>> https://peerj.com/preprints/2381/
an implementation of the proposed specification of the “Graphical Fragment Assembly” (GFA) format in the Ruby programming language. antisense. @razoralign 5分5分前 翻訳を表示
If a segment end e has a single link l, connecting it to a segment end e’, l must be present in any Hamiltonian path (it is mandatory). RGFATools provides a method which detects all mandatory links in the graph and removes all superfluous links. enforced mandatory links and randomly oriented invertible segments in a loop, merging linear paths after each operation.
gfa =" RGFA.from_file("graph.gfa")
gfa.each_segment{|s| puts(s. name + "\t" + s.length)}
□ How DNA could store all the world’s data:
>> http://www.nature.com/news/how-dna-could-store-all-the-world-s-data-1.20496
□ A Mathematical Framework for Feature Selection from Real-World Data with Non-Linear Observations:
>> http://arxiv.org/pdf/1608.08852v1.pdf
A major difficulty is that these variables usually cannot be observed directly but rather arise as hidden factors in the actual data vectors. construct an optimal representation of the signal vector in order to mimic the original problem of variable selection within the data domain. it has turned out that this approach works almost as good as if one would explicitly know the hidden signal factors of “oracle property”.

□ Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model:
>> http://biorxiv.org/content/biorxiv/early/2016/09/03/073239.full.pdf
a new deep learning method for contact prediction that predicts contacts by integrating both evolutionary coupling (EC) information and sequence conservation information through an ultra-deep neural network consisting of two deep residual neural networks. dramatically improves contact prediction, exceeding currently the best methods CCMpred, Evfold, PSICOV & MetaPSICOV by a very large margin.
□ SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences
>> http://biorxiv.org/content/biorxiv/early/2016/09/09/074161.full.pdf
SINTAX achieves comparable or better accuracy to the RDP Naive Bayesian Classifier with a simpler algorithm that does not require training. Assuming 47% novel genera and the lower OC value gives an estimate of 0.47×0.40×3.2M = 600k over-classified genera.
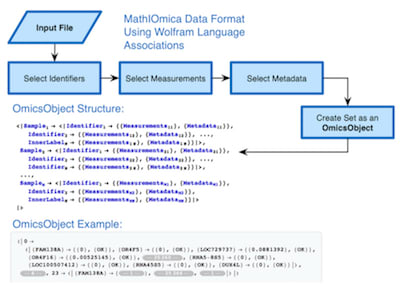
□ MathIOmica: An Integrative Platform for Dynamic Omics in the Wolfram Language:
>> http://biorxiv.org/content/biorxiv/early/2016/09/10/074260.full.pdf
The main functionality exists to use a Lomb-Scargle transformation to handle uneven sampling and/or missing data, an approach that has been adapted from astronomy and used in the analysis and classification of dynamics in biological systems.
□ ENVIREM: An expanded set of bioclimatic and topographic variables increases flexibility:
>> http://biorxiv.org/content/biorxiv/early/2016/09/14/075200.full.pdf
when knowledge about the determinants of species distributions is available from ecological theory, the ENVIREM variables may be particularly useful for developing and testing the predictions of species-specific hypotheses.
□ Using Nonlinearity in Understanding Market Forces:
>> http://scitation.aip.org/content/aip/journal/chaos/26/9/10.1063/1.4962296
a dynamic discrete-time theoretical model where managerial price-setting firms design contracts based on relative performances. A possible extension is that of studying heterogeneous behaviour of players in models of Bertrand rivalry by considering one player with limited information and Local Monopolistic Approximation and the other player with full information and naïve expectations.

□ "Metaheuristics for String Problems in Bio-informatics" ー 線形計画法のバイオインフォマティクス応用からの導入
□ 必要に迫られて相対論的ナビエ・ストークスについて2日でマスターしなければならない。 A covariant action principle for dissipative fluid dynamics: http://arxiv.org/pdf/1306.3345v1.pdf
EMテンソルとは、実事象の何を表現するのか。これは『観察者が何の夢を見ているか』の定義に置き換えられる。単位時空を透過する粒子密度が散逸的なら、それが運動量の差異として観測される。然し密度の相対性とは、グリッドの粗視化によっても齎される。ビートの間隔が狭まるほど「速く」感じる。
□ FBB: A Fast Bayesian Bound tool to calibrate RNA-seq aligners:
>> https://bitbucket.org/irenerodriguez/fbb
a Fast Bayesian Bound (FBB) that serves as a canonical reference to compare alignment results across different algorithms.
□ Final version of Graphical Fragment Assembly (GFA) Format Specification 1.0 is tagged, anticipating upcoming GFA 2.0
>> https://github.com/GFA-spec/GFA-spec
□ Nanonet: Nanonet provides recurrent neural network basecalling for Oxford Nanopore MinION data:
>> https://github.com/nanoporetech/nanonet
Nanonet contains implementations of both 1D and 2D basecalling with OpenCL. only the canonical 1D basecalling library will be compiled; Nanonet contains a squiggle simulator based on a similar Markov model to that which can be used for basecalling. A base sequence is first converted to a sequence of overlapping kmers and is currupted by movement artifacts.
□ nanonet basecaller 2D available in bioconda:
>> https://bioconda.github.io/recipes/nanonet/README.html
Nanonet provides recurrent neural network basecalling for Oxford Nanopore MinION data.
BOOST_PYTHON_MODULE(viterbi_2d) {
import_array();
bp::numeric::array::set_module_and_type("numpy", "ndarray");
□ Nanopore DNA Sequencing and Genome Assembly on the International Space Station:
>> http://biorxiv.org/content/early/2016/09/27/077651
□ SAMSA: a comprehensive metatranscriptome analysis pipelinewhich runs in conjunction with Metagenome-RAST (MG-RAST):
>> http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1270-8
To evaluate necessary sequencing depth using a bioinformatics based approach, generated 100 randomly selected subsets of a large and comparatively over-sequenced metatranscriptome of 21.6 million annotated reads (derived from 38 M raw reads), creating ten smaller stand-alone simulated metatranscriptomes for each size point measured, moving in ten percent increments from 1 million up to 20 annotated million reads per subset.









※コメント投稿者のブログIDはブログ作成者のみに通知されます