









□ We simply cannot go on being so vague about ‘function’:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1600-4
Philosophers have also pointed out that ecologists, physiologists, and molecular biologists and genomicists tend to be satisfied with ‘what it does’ or causal role explanations, whereas evolutionary biologists also require ‘why it’s there’ or selected effect rationales. it is clearly wrong to use conclusions based on one to ‘refute’ hypotheses based on the other.
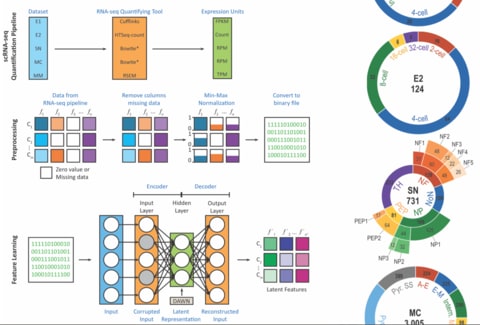
□ DUSC: A Hybrid Deep Clustering Approach for Robust Cell Type Profiling Using Single-cell RNA-seq Data:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/04/511626.full.pdf
a novel extension to the DAE called Denoising Autoencoder With Neuronal approximator (DAWN), which decides the number of latent features that is required to represent efficiently any given dataset. using the features generated by DAWN as an input to the EM clustering algorithm and show that the hybrid deep clustering approach, DUSC (Deep Unsupervised Single-cell Clustering), has higher accuracy.
□ Hybrid assembly of ultra-long nanopore reads augmented with 10×-genomics contigs: Demonstrated with a human genome:
>> https://www.sciencedirect.com/science/article/pii/S0888754318305603
demonstrate the feasibility of integrating the 3GS with 10×-Genomics technologies for a new strategy of hybrid de novo genome assembly by utilizing DBG2OLC and Sparc software packages.
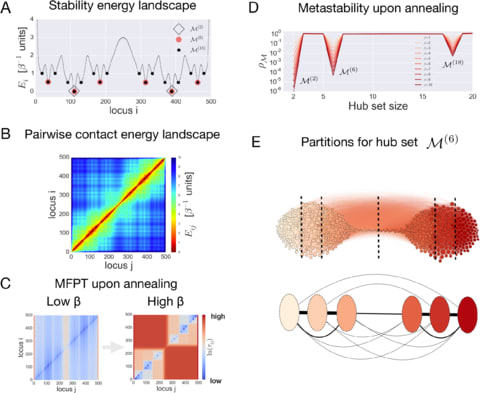
□ ChromaWalker: Exploring chromatin hierarchical organization via Markov State Modelling:
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1006686
A Markov process describes the random walk of a traveling probe in the corresponding energy landscape, mimicking the motion of a biomolecule involved in chromatin function. By studying the metastability of the associated Markov State Model upon annealing, the hierarchical structure of individual chromosomes is observed, and a corresponding set of structural partitions is identified at each level of hierarchy.

□ OGRE: Overlap Graph-based metagenomic Read clustEring:
>> https://www.biorxiv.org/content/early/2019/01/03/511014
OGRE employs Minimap2, a heuristic approach for the efficient construction of an overlap graph. but the single linkage algorithm is sequential and has a complexity of O(n log n+n), applying it to the long edge list obtained results in unacceptable computation times. About half of these overlaps could be filtered out using a logistic regression on the overlap length and a Phred-based matching probability.

□ B-LORE: Bayesian multiple logistic regression for case-control GWAS:
>> https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1007856
B-LORE, a scalable Bayesian method for multiple logistic regression. the quasi-Laplace approximation to solve the integral and avoid MCMC sampling. B-LORE is a command line tool that creates summary statistics from multiple logistic regression on GWAS data, and combines the summary statistics from multiple studies in a meta-analysis.
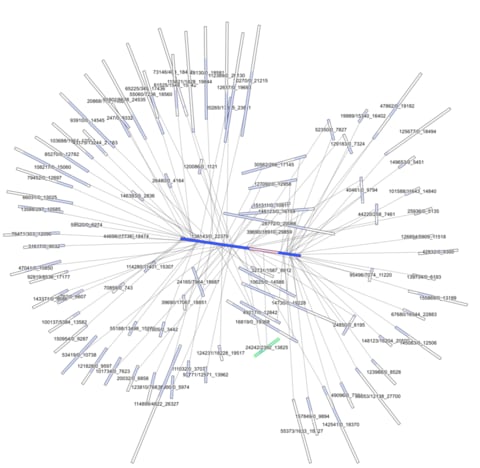
□ GfaViz: Flexible and interactive visualization of GFA sequence graphs:
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/bty1046/5267826
the Graphical Fragment Assembly 2 (GFA2) format introduces several features, which makes it suitable for representing other kinds of information, such as scaffolding graphs, variation graphs, alignment graphs, and colored metagenomic graphs. The visualizations can be exported to raster and vector graphics formats.
The assembly graph of the scaffold sequences is output as GFA 2 by the Abyss assembler when using the option graph=gfa2. This graph contains dovetail overlaps between pairs of scaffolds, but has no lines describing the gaps. Thus, to obtain a complete scaffolding graph, the abyss-to-dot tool with the options--gfa2 --estimate was applied to the DOT file containing distance estimates output by abyss-scaffold.
□ LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly:
>> https://academic.oup.com/gigascience/advance-article/doi/10.1093/gigascience/giy157/5256637
LR_Gapcloser is flexible in that both Pacbio reads and Nanopore reads can be utilized to fill the gaps, and also likely to use the 10X genomic linked reads and pre-assembled contigs to fill gaps in the assemblies.

□ NCLcomparator: systematically post-screening non-co-linear transcripts (circular, trans-spliced, or fusion RNAs) identified from various detectors:
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-018-2589-0
NCLcomparator first examine whether the input Non-co-linear (NCL) events are potentially false positives derived from ambiguous alignments (i.e., the NCL events have an alternative co-linear explanation or multiple matches against the reference genome).
□ Mapping and characterization of structural variation in 17,795 deeply sequenced human genomes:
>> https://www.biorxiv.org/content/biorxiv/early/2018/12/31/508515.full.pdf
□ BioSAILs: versatile workflow management for high-throughput data analysis:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/02/509455.full.pdf
BioSAILs (Bioinformatics Standardized Analysis Information Layers), a scientific workflow management system (WMS) comprises two central components, BioX command and HPCRunner command, supported by BioStacks software stacks.
□ Multi-CSAR: a multiple reference-based contig scaffolder using algebraic rearrangements:
>> https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-018-0654-y
utilize a heuristic method to develop a new scaffolder called Multi-CSAR that is able to accurately scaffold a target draft genome based on multiple reference genomes, each of which does not need to be complete. the experimental results on real datasets show that Multi-CSAR outperforms other multiple reference-based scaffolding tools, Ragout and MeDuSa, in terms of many average metrics.

□ Selective single molecule sequencing and assembly of a human Y chromosome of African origin:
>> https://www.nature.com/articles/s41467-018-07885-5
a novel strategy to sequence native, unamplified flow sorted DNA on a MinION nanopore sequencing device. It constitutes a significant improvement over comparable previous methods, increasing continuity by more than 800%.
□ "Algorithms" by Jeff Erickson
>> http://jeffe.cs.illinois.edu/teaching/algorithms/
the definition of n-beat meters in Sanskrit was one of the earliest examples of recursion. The Fibonacci numbers originated in Indian poetry, many centuries before Fibonacci.
□ "Causal Inference Book" by Miguel Hernan
>> https://www.hsph.harvard.edu/miguel-hernan/causal-inference-book/
The book is organized in 3 parts of increasing difficulty: From counterfactuals and causal diagrams to treatment-confounder feedback and g-methods.
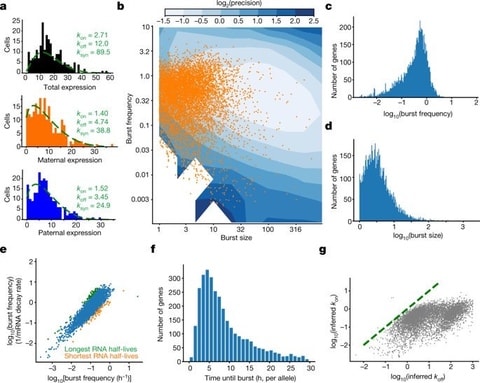
□ Genomic encoding of transcriptional burst kinetics:
>> https://www.nature.com/articles/s41586-018-0836-1
the core promoter elements affect burst size and uncover synergistic effects between TATA and initiator elements, which were masked at mean expression levels. burst frequency is primarily encoded in enhancers and burst size in core promoters, and that allelic single-cell RNA sequencing is a powerful model for investigating transcriptional kinetics.
□ "Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems" accepted by IPDPS'19
>> http://www.ipdps.org
□ A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens
>> https://www.cell.com/cell/fulltext/S0092-8674(18)31554-X
a genome-wide framework for enhancer-gene pairings via multiplex CRISPRi + scRNA-seq. "the assay formerly known as crisprQTL", attempt at-scale validation and pairing of candidate regulatory elements with their target genes.

□ The dynamics of adaptive genetic diversity during the early stages of clonal evolution:
>> https://www.nature.com/articles/s41559-018-0758-1
a crash in adaptive diversity follows, caused by highly fit double-mutant ‘jackpot’ clones that are fed from exponentially growing single mutants, a process closely related to the classic Luria–Delbrück experiment. The diversity crash is likely to be a general feature of asexual evolution with clonal interference; however, both its timing and magnitude are stochastic and depend on the population size, the distribution of beneficial fitness effects and patterns of epistasis.
□ plyranges: a grammar of genomic data transformation:
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1597-8
the Genome Query Language (GQL) and its distributed implementation GenAp which use a SQL-like syntax for fast retrieval of information of unprocessed sequencing data. Similarly, the Genometric Query Language (GMQL) implements a DSL for combining genomic datasets.
a genomic DSL called plyranges that reformulates notions from existing genomic algebras and embeds them in R as a genomic extension of dplyr. By analogy, plyranges is to the genomic algebra, as dplyr is to the relational algebra.

□ An updated and significantly expanded version of the "UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction":
>> https://arxiv.org/abs/1802.03426
More explanation, algorithm descriptions, and more experiments looking at stability, and working directly on high dimensional data -- as high as 1.8 million dimensional data.

□scSVA: an interactive tool for big data visualization and exploration in single-cell omics:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/06/512582.full.pdf
scSVA is memory efficient for more than hundreds of millions of cells, can be run locally or in a cloud, and generates high-quality figures. scSVA is a numerically efficient method for single-cell data embedding in 3D which combines an optimized implementation of diffusion maps with a 3D force-directed layout, enabling generation of 3D data visualizations at the scale of a million cells.

□ LEARNA: Learning to Design RNA using a deep reinforcement learning algorithm:
>> https://arxiv.org/pdf/1812.11951.pdf
a new algorithm for the RNA Design problem, dubbed LEARNA. LEARNA uses deep reinforcement learning to train a policy net- work to sequentially design an entire RNA sequence given a specified secondary target structure.
Meta-LEARNA constructs an RNA Design policy that can be applied out of the box to solve novel RNA target structures by meta-learning across 8000 different structures for one hour on 20 cores.
□ End-to-End Differentiable Physics for Learning and Control:
>> https://papers.nips.cc/paper/7948-end-to-end-differentiable-physics-for-learning-and-control.pdf
demonstrate how to perform backpropagation analytically through a physical simulator defined via a linear complementarity problem. Through experiments in diverse domains, highlight the system’s ability to learn physical parameters from data, efficiently match and simulate observed visual behavior, and readily enable control via gradient-based planning methods.
a deep learning wrapper around a differentiable physics engine and then can rapidly learn to fix the errors of the physics engine. treat a deep neural network output as an energy function/probability space (with appropriate marginalisation).
□ 10X Genomics raises $35 million for cell sequencing:
>> https://venturebeat.com/2019/01/07/10x-genomics-35-million-series-d-funding/
The global genomics market, according to Grand View Research, it’s forecast to be worth $27.6 billion, led by pioneers such as Verge Genomics, Sophia Genetics, and Shivom. 10X Genomics develops a suite of gene analysis tools, today announced it has secured $35 million in financing in an extension of its April Series D. The round, led by Meritech Capital, with participation from Wells Fargo and Fidelity, brings 10X’s total raised to $243 million.
□ Bio-Rad Laboratories Inc. and the University of Chicago have won a $24 million patent infringement verdict against gene-sequencing startup 10x Genomics Inc.
>> https://finance.yahoo.com/news/bio-rad-snares-24-million-072407019.html

□ The DNA Walk and its Demonstration of Deterministic Chaos:
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/bty1021/5280132
Using fractal analysis for the DNA walks, determined the complexity and nucleotide variance of commonly observed mutated genes, and their wild type counterparts. DNA walks for wild type genes demonstrate varying levels of chaos.

□ Central dogma rates and the trade-off between precision and economy in gene expression:
>> https://www.nature.com/articles/s41467-018-07391-8
explain the empty Crick space by a trade-off between cost and noise of gene expression. This theory accurately predicts the boundary of the empty region which varies by 2 orders of magnitude between the model organisms they considered.
□ ELECTOR: Evaluator for long reads correction methods:
>> https://www.biorxiv.org/content/biorxiv/early/2019/01/07/512889.full.pdf
ELECTOR is an efficient segmentation heuristic for multiple alignment. this tool processes the different fragments separately before reconstituting the whole alignment, and takes into account missing parts.

□ SAVER-X: Transfer learning in single-cell transcriptomics improves data denoising and pattern discovery:
>> https://www.biorxiv.org/content/biorxiv/early/2018/11/01/457879.full.pdf
SAVER-X uses the deep autoencoder, a neural network that achieves noise reduction by means of an information bottleneck. The deep learning model in SAVER-X extracts transferable gene expression features across data from different labs, generated by varying technologies, and obtained from divergent species.
□ Parallelized Natural Extension Reference Frame: Parallelized Conversion from Internal to Cartesian Coordinates:
>> https://www.ncbi.nlm.nih.gov/pubmed/30614534
A mathematically equivalent algorithm, pNeRF, has been derived that is parallelizable along a polymer's length. In machine learning-based workflows, in which partial derivatives are backpropagated through NeRF equations and neural network primitives, switching to pNeRF can reduce the fractional computational cost of coordinate conversion from over two-thirds to around 10%.
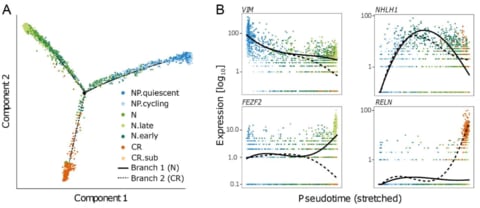
□ CellSIUS provides sensitive and specific detection of rare cell populations from complex single cell RNA-seq data:
>> https://www.biorxiv.org/content/early/2019/01/09/514950
exemplify the use of CellSIUS for the characterization of a human pluripotent cell 3D spheroid differentiation protocol recapitulating deep-layer corticogenesis in vitro.

□ The harmonic mean p-value for combining dependent tests:
>> https://www.pnas.org/content/early/2019/01/08/1814092116
the harmonic mean p-value (HMP), a simple to use and widely applicable alternative to Bonferroni correction motivated by Bayesian model averaging. The power of the HMP to detect significant hypothesis groups is greater than the power of the Benjamini–Hochberg procedure to detect significant hypotheses, although the latter only controls the weaker false discovery rate (FDR).









