










□ Storing and analyzing a genome on a blockchain
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02699-7
Nebula Genomics uses Ethereum Smart Contracts to facilitate communication between nodes, and Blockstack to facilitate data storage, but Blockstack stores the data off-chain, either on a local drive or in the cloud.
SAMchain is the first framework to store raw genomic reads on a blockchain, on-chain. The algorithm searches through the binned streams to obtain the SAM data. Each private blockchain network corresponds to a single genome owned by the individual to which the genome belongs.
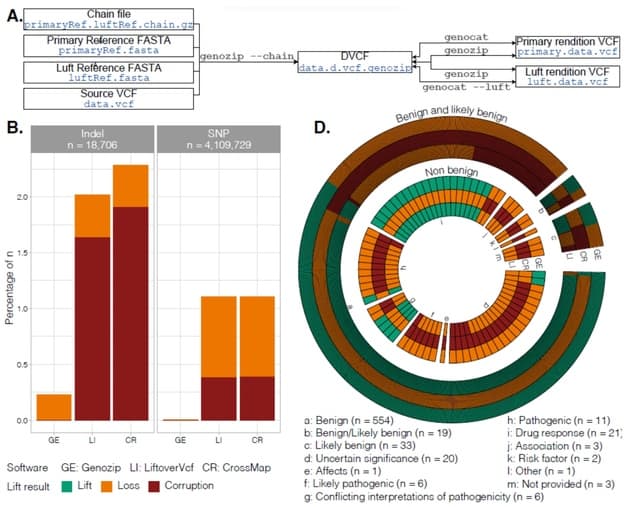
□ Genozip Dual-Coordinate VCF format enables efficient genomic analyses and alleviates liftover limitations
>> https://www.biorxiv.org/content/10.1101/2022.07.17.500374v1.full.pdf
Dual Coordinate VCF (DVCF), a file format that records genomic variants against two different reference genomes simultaneously and is fully compliant with the current VCF specification.
Using DVCF files, researchers can alternate between coordinate systems according to their needs – without creating duplicate VCF files. Importantly the DVCF file format is independent of its implementation in Genozip.
□ PolarMorphism enables discovery of shared genetic variants across multiple traits from GWAS summary statistics
>> https://academic.oup.com/bioinformatics/article/38/Supplement_1/i212/6617483
PolarMorphism, a new approach to identify pleiotropic SNPs that is more efficient, identifies the same number of pleiotropic SNPs as PLACO, but can be applied to more than two traits. This enables the identification of SNPs that have an effect on numerous traits.
PolarMorphism enables construction of a trait network showing which traits share SNPs. PolarMorphism identifies more pleiotropic SNPs than the standard intersection method and than PRIMO. PolarMorphism finished analysis of 1 million SNPs in less than 20 s.
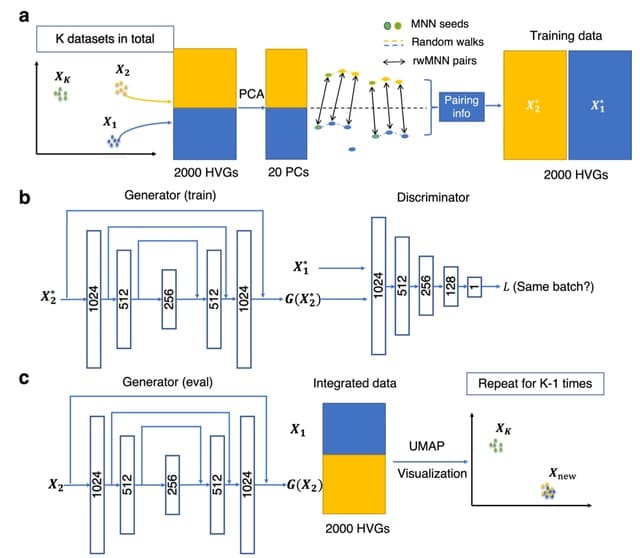
□ ResPAN: a powerful batch correction model for scRNA-seq data through residual adversarial networks
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac427/6623406
ResPAN is a light structured Residual autoencoder and mutual nearest neighbor Paring guided Adversarial Network for scRNA-seq batch correction.
ResPAN is based on Wasserstein Generative Adversarial Network (WGAN) combined with random walk mutual nearest neighbor pairing and fully skip-connected autoencoders to reduce the differences among batches.
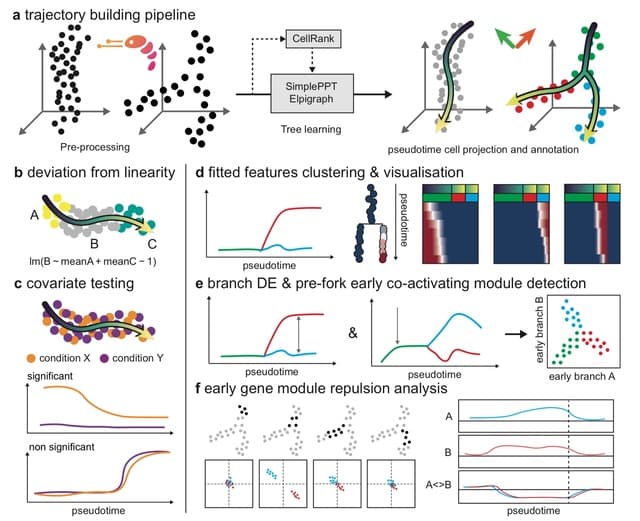
□ scFates: a scalable python package for advanced pseudotime and bifurcation analysis from single cell data
>> https://www.biorxiv.org/content/10.1101/2022.07.09.498657v1.full.pdf
scFates is fully compatible with scanpy ecosystem by using the anndata format, and provides GPU and multicore accelerated functions for faster and more scalable inference.
Using SimplePPT algorithm, where each cell is assigned a probability to each principal point, scFates can generate several pseudotime mappings. scFates provides functions for selecting specific portions of the tree, by selecting starting and endpoints, or by using pseudotime.
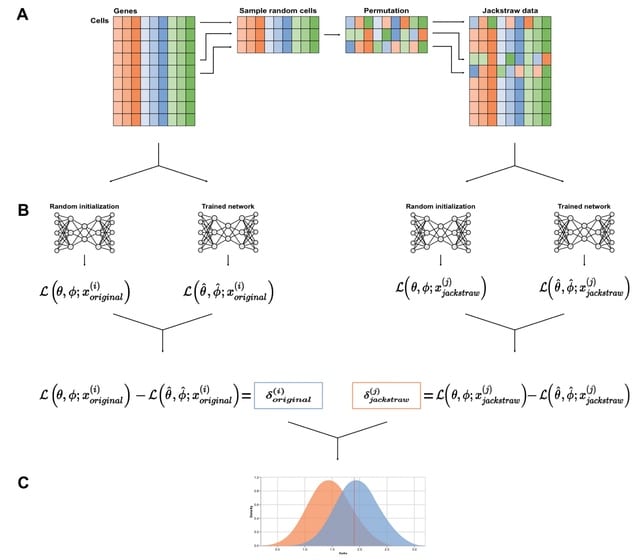
□ scVIDE: Designing Single-Cell RNA-Sequencing Experiments for Learning Latent Representations
>> https://www.biorxiv.org/content/10.1101/2022.07.08.499284v1.full.pdf
scVIDE determines statistical power for detecting cell group structure in a lower-dimensional representation. scVIDE starts with a cell by gene count matrix from which a small number of cells are randomly selected and counts are randomly permuted across genes.
Eextending scVIDE to deep Boltzmann machines (DBMs), which have been adapted to scRNA-seq data, could be useful because it was previously shown that DBMs could learn from smaller data sets compared to other deep generative models.

□ scDLC: a deep learning framework to classify large sample single-cell RNA-seq data
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08715-1
scDLC is based on the long short-term memory recurrent neural networks (LSTMs). This classifier does not require a prior knowledge on the scRNA-seq data distribution and it is a scale invariant method which does not require a normalization procedure for scRNA-seq data.
scDLC amplifies the features of the selected genes through the first fully connected layer. The output of the 1st fully connected layer is taken as the input of the two-layer long short-term memory network layer, and the weights of all gates are estimated by network calculation.

□ Deep Visualization: Structure-Preserving and Batch-Correcting Visualization Using Deep Manifold Transformation for Single-cell RNA-Seq Profiles
>> https://www.biorxiv.org/content/10.1101/2022.07.09.499435v1.full.pdf
deep visualization (DV), that possesses the ability to preserve inherent structure of data and handle batch effects and is applicable to a variety of datasets from different application domains and dataset scales.
The method embeds a given dataset into a 2- or 3-dimensional visualization space, with either a Euclidean or hyperbolic metric depending on a specified task type and data type “time-fixed” and “time-evolution” scRNA-seq data, respectively.
DV learns a semantic graph to describe the relationships between data samples, transforms the data into visualization space while preserving the geometric structure of the data and correcting batch effects in an end-to-end manner.
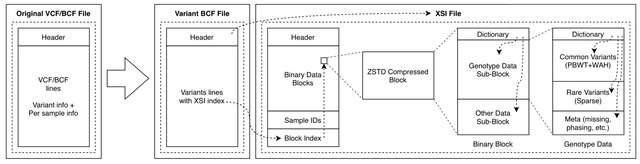
□ XSI - A genotype compression tool for compressive genomics in large biobanks
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac413/6617346
xSqueezeIt (XSI) - VCF / BCF Genotype Compressor based on sparse representation for rare variants and positional Burrows-Wheeler transform (PBWT) followed by 16-bit Word Aligned Hybrid (WAH) encoding for common variants.
XSI relies on a hierarchical block-based strategy. The blocks hold a small dictionary referencing their content. The Sub-blocks are compressed with specific to the data type. The PBWT is recomputed from the initial sample ordering for each block, making each block independent.
□ The Practical Haplotype Graph, a platform for storing and using pangenomes for imputation
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac410/6617344
The Practical Haplotype Graph is a pangenome pipeline, database (PostGRES & SQLite), data model (Java, Kotlin, or R), and Breeding API (BrAPI). At even 0.1X coverage, with appropriate reads and sequence alignment, imputation results in extremely accurate haplotype reconstruction.
The Practical Haplotype Graph is a trellis graph that represents discrete genomic DNA sequences and connections. HMM algorithms, Viterbi and forward-backward, operate on a trellis graph, and organize pangenomes by aligning all of the genomes against a single reference genome.
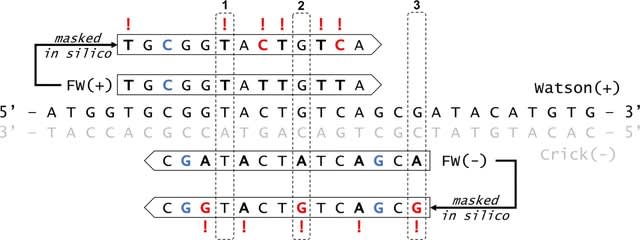
□ Revelio: Manipulating base quality scores enables variant calling from bisulfite sequencing alignments using conventional bayesian approaches
>> https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08691-6
The double-masking procedure facilitates sensitive and accurate variant calling directly from bisulfite sequencing data using software intended for conventional DNA sequencing libraries.
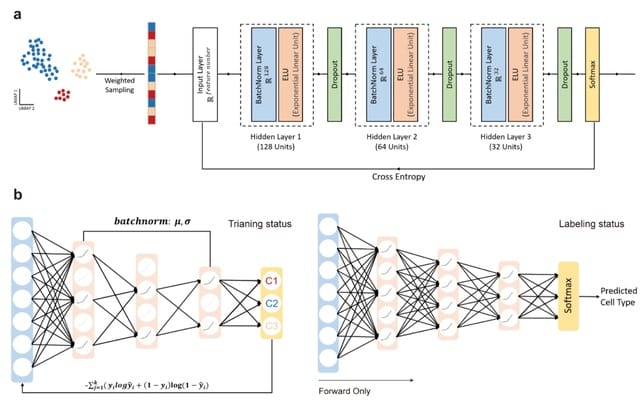
□ scBalance: A scalable sparse neural network framework for rare cell type annotation of single-cell transcriptome data
>> https://www.biorxiv.org/content/10.1101/2022.06.22.497193v1.full.pdf
scBalance, a sparse neural network framework to automatically label rare cell types in all scale scRNA-seq datasets. By leveraging the newly designed neural network structure, scBalance especially obtains an outperformance on rare cell type annotation and robustness on batch effect.
scBalance leverages the combination of weight and sparse neural network, whereby rare cell types are informative w/o harming the annotation efficiency of the major cell populations. scBalance is the first auto-annotation tool that expands scalability to 1.5 million cells dataset.
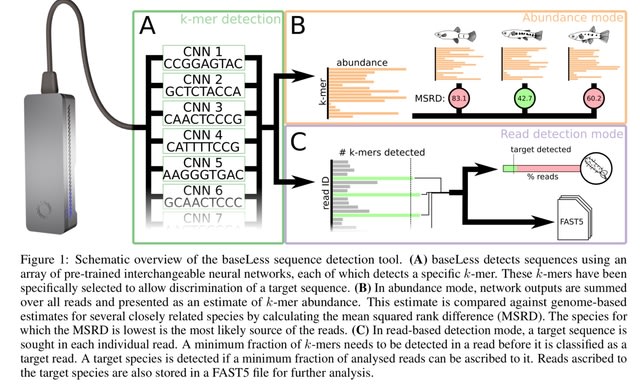
□ baseLess: Lightweight detection of sequences in raw MinION data
>> https://www.biorxiv.org/content/10.1101/2022.07.10.499286v1.full.pdf
baseLess, a computational tool that enables such target-detection-only analysis. BaseLess makes use of an array of small neural networks, each of which efficiently detects a fixed-size subsequence of the target sequence directly from the electrical signal.
baseLess baseLess deduces the presence of a target sequence by detecting squiggle segments corresponding to salient short sequences, k-mers, using an array of convolutional neural networks.
baseLess ranks k-mers by abundance as measured in the reads and compares it to their abundance ranking in the target and background genomes, using the mean squared rank difference (MSRD).
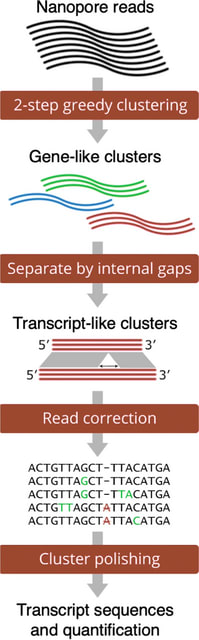
□ RATTLE: reference-free reconstruction and quantification of transcriptomes from Nanopore sequencing
>> https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02715-w
RATTLE is competitive at recovering transcript sequences and their abundances despite not using any information from the reference. RATTLE lays the foundation for a multitude of potential new applications of Nanopore transcriptomics.
RATTLE performs a greedy deterministic clustering using a two-step k-mer based similarity measure. RATTLE solves the Longest Increasing Subsequence (LIS) problem to find the longest list of collinear matching k-mers between a pair of reads.
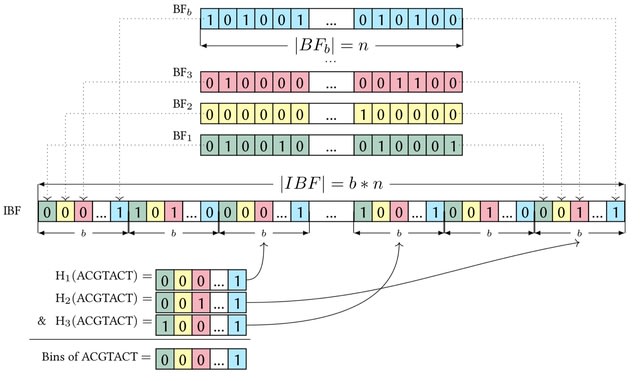
□ Needle: A fast and space-efficient prefilter for estimating the quantification of very large collections of expression experiments
>> https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btac492/6633930
Needle, a fast and space-efficient prefilter for estimating the quantification of very large nucleotide sequences. Needle can estimate the quantification of thousands of sequences in a few minutes or even only seconds.
Needle uses the Interleaved Bloom Filter (IBF) with minimizers instead of contiguously overlapping k-mers to efficiently index and query these experiments. Needle splits the count values of one experiment into meaningful buckets and stores each bucket as one IBF.

□ HiCImpute: A Bayesian hierarchical model for identifying structural zeros and enhancing single cell Hi-C data
>> https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010129
HiCImpute, a Bayesian hierarchical model that goes beyond data quality improvement by also identifying observed zeros that are in fact structural zeros.
The key idea relies on the introduction of an indicator variable (the latent variable) denoting structural zeros or otherwise, for which a statistical inference is made based on its posterior probability estimated using Markov chain Monte Carlo (MCMC) samples.
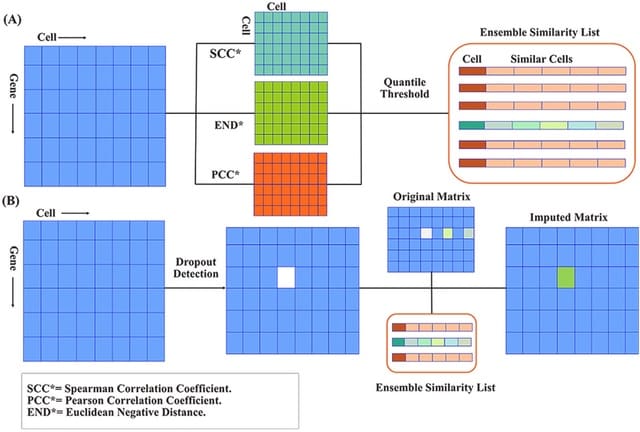
□ CDSImpute: An ensemble similarity imputation method for single-cell RNA sequence dropouts
>> https://www.sciencedirect.com/science/article/abs/pii/S0010482522004504
CDSImpute (Correlation Distance Similarity Imputation), a novel Single-cell RNA dropout imputation method to retrieve the original gene expression of the genes with excessive zero and near-zero counts.
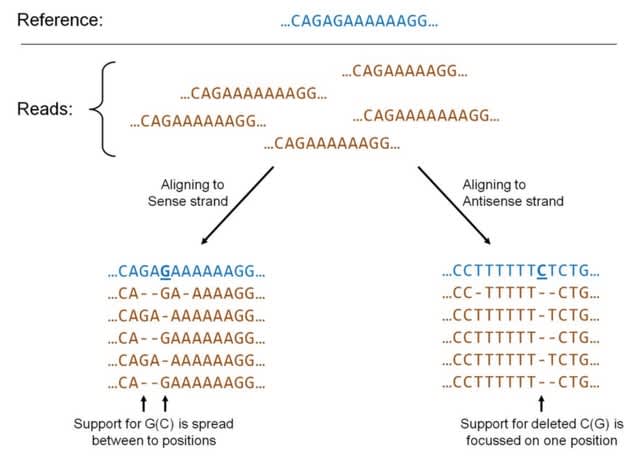
□ Complete sequence verification of plasmid DNA using the Oxford Nanopore Technologies′ MinION device
>> https://www.biorxiv.org/content/10.1101/2022.06.21.497051v1.full.pdf
A pipeline that generates a high-quality consensus sequence of linearized plasmid using ONT MinION sequencing, leveraging substantial sequencing depth and stringent quality filters to overcome the relatively high error rates associated with nanopore sequencing.

□ Mandalorion: Identifying and quantifying isoforms from accurate full-length transcriptome sequencing reads
>> https://www.biorxiv.org/content/10.1101/2022.06.29.498139v1.full.pdf
The Mandalorion tool is continuously developed over the last 5 years, identifies and quantifies high-confidence isoforms from accurate full-length transcriptome sequencing reads produced by methods like PacBio Iso-Seq and ONT-based R2C2.
Mandalorion v4 accepts an arbitrary number of fasta/q files containing accurate full-length transcriptome sequencing data. Mandalorion v4 identifies isoforms with very high Recall and Precision when applied to either spike-in or simulated data with known ground-truth isoforms.
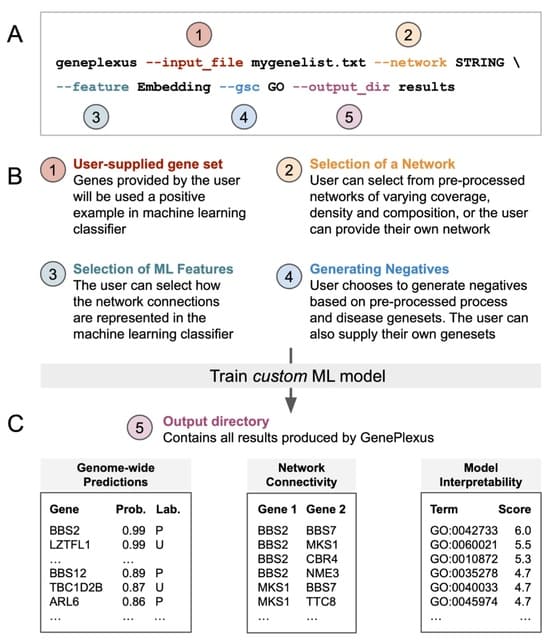
□ PyGenePlexus: A Python package for gene discovery using network-based machine learning
>> https://www.biorxiv.org/content/10.1101/2022.07.02.498552v1.full.pdf
The GenePlexus method utilizes pre-processed information from genome-wide molecular networks and gene set collections from the Gene Ontology (GO) and DisGeNet.
PyGenePlexus trains a custom ML model and returns the probability of how associated every gene in the network is to the user supplied gene set, along with the network connectivity of the top predicted genes.
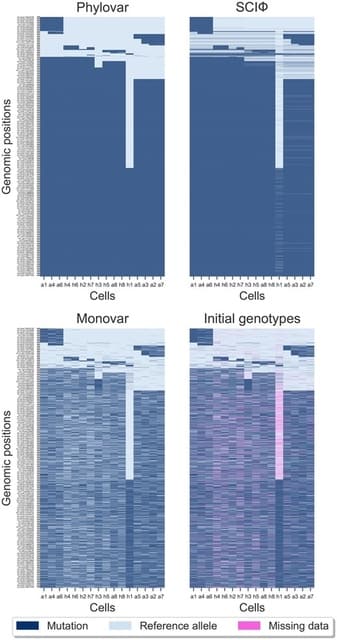
□ Phylovar: toward scalable phylogeny-aware inference of single-nucleotide variations from single-cell DNA sequencing data
>> https://academic.oup.com/bioinformatics/article/38/Supplement_1/i195/6617481
Phylovar, which extends the phylogeny-guided variant calling approach to sequencing datasets containing millions of loci. Phylovar outperforms SCIΦ
in terms of running time while being more accurate than Monovar in terms of SNV detection.
Phylovar finds the tree topology and the placement of mutations on ancestral single cells that maximize the likelihood of the erroneous observed read counts given the genotypes.
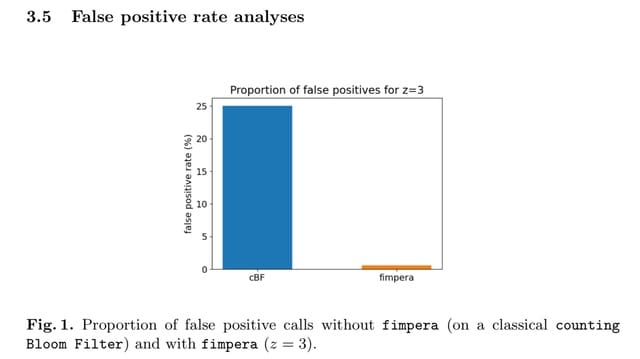
□ fimpera: drastic improvement of Approximate Membership Query data-structures with counts
>> https://www.biorxiv.org/content/10.1101/2022.06.27.497694v1.full.pdf
fimpera, consisting of a simple strategy for reducing the false-positive rate of any AMQ indexing all k-mers (words of length k) from a set of sequences, along with their abundance information.
fimpera decreases the false-positive rate of a counting Bloom filter by an order of magnitude while reducing the number of overestimated calls, as well as lowering the average difference between the overestimated calls and the ground truth.
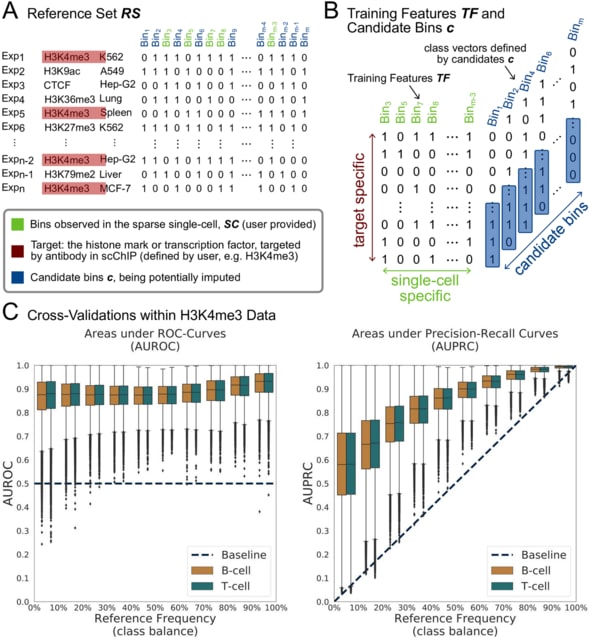
□ SIMPA: Single-cell specific and interpretable machine learning models for sparse scChIP-seq data imputation
>> https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0270043
SIMPA, a scChIP-seq data imputation method leveraging predictive information within bulk data from the ENCODE project to impute missing protein-DNA interacting regions of target histone marks or transcription factors.
SIMPA enables the interpretation of machine learning models by revealing interaction sites of a given single cell that are most important for the imputation model trained for a specific genomic region.
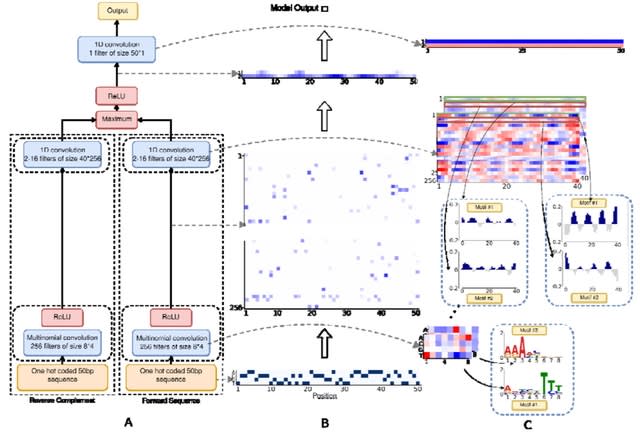
□ DeepBend: An Interpretable Model of DNA Bendability
>> https://www.biorxiv.org/content/10.1101/2022.07.06.499067v1.full.pdf
DeepBend, a convolutional neural network model built as a visible neural network where we designed the convolutions to directly capture the motifs underlying DNA bendability and how their periodic occurrences or relative arrangements modulate bendability.
DeepBend is a 3-layered CNN that takes in a one-hot encoded DNA sequence as input and predicts its bendability. Each row of a first layer filter is a multinomial distribution over the four nucleotides, these filters are interpretable as biophysical models of sequence motifs.
□ DeepGenGrep: a general deep learning-based predictor for multiple genomic signals and regions
>> https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/btac454/6633307
DeepGenGrep, a general predictor for the systematic identification of multiple different GSRs from genomic DNA sequences.
DeepGenGrep leverages the power of hybrid neural networks comprising a three-layer convolutional neural network and a two-layer long short-term memory to effectively learn useful feature representations from sequences.

□ pareg: Coherent pathway enrichment estimation by modeling inter-pathway dependencies using regularized regression
>> https://www.biorxiv.org/content/10.1101/2022.07.06.498967v1.full.pdf
pareg follows the ideas of GSEA as it requires no stratification of the input gene list, of MGSA as it incorporates term-term relations in a database-agnostic way, and of LRPath as it makes use of the flexibility of the regression approach.
pareg assumes that a linear combination of gene-pathway memberships is driving the overall pathway dysregulation, an assumption which may reduce the algorithm’s applicability in certain biological environments.

□ Porechop_ABI: discovering unknown adapters in ONT sequencing reads for downstream trimming
>> https://www.biorxiv.org/content/10.1101/2022.07.07.499093v1.full.pdf
Porechop_ABI automatically infers adapter sequences from raw reads alone, without any external knowledge or database. This algorithm determines whether the reads contain adapters, and if so what the content of these adapters is.
Porechop_ABI uses technics coming from string algorithms, with approximate k-mer, full text indexing and assembly graphs. Porechop_ABI cleans untrimmed reads for which the adapter sequences are not documented, to check whether a dataset has been trimmed or not.
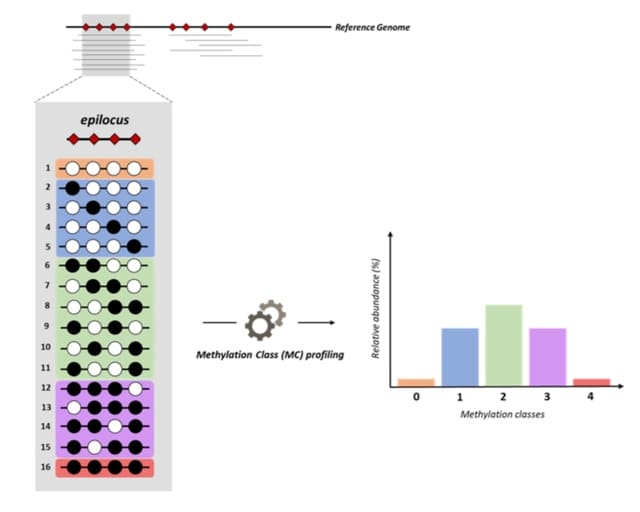
□ MC profiling: a novel approach to analyze DNA methylation heterogeneity in genome-wide bisulfite sequencing data
>> https://www.biorxiv.org/content/10.1101/2022.07.06.498979v1.full.pdf
Methylation Class (MC) profiling approach is built on the concept of MCs, i.e. groups of DNA molecules sharing the same number of methylated cytosines in a sample.
MC profiling identified cell-to-cell differences as the prevalent contributor to DNA methylation heterogeneity, with allele differences emerging in a small fraction of analyzed regions. Moreover, MC profiling led to the identification of signatures of loci undergoing genomic imprinting.

□ MINE is a method for detecting spatial density of regulatory chromatin interactions based on a MultI-modal NEtwork
>> https://www.biorxiv.org/content/10.1101/2022.07.11.499656v1.full.pdf
MINE-Loop is a neural network model that integrates Hi-C, ChIP-seq, and ATAC-seq data to enhance the proportion of detectable regulatory chromatin interactions by reducing noise from non-regulatory interactions.
MINE-Density can be used to calculate the spatial density of regulatory chromatin interactions (SD-RCI) identified by MINE-Loop, and MINE-Viewer facilitates visualization of density and specific interactions with regulatory factors in 3D genomic structures.
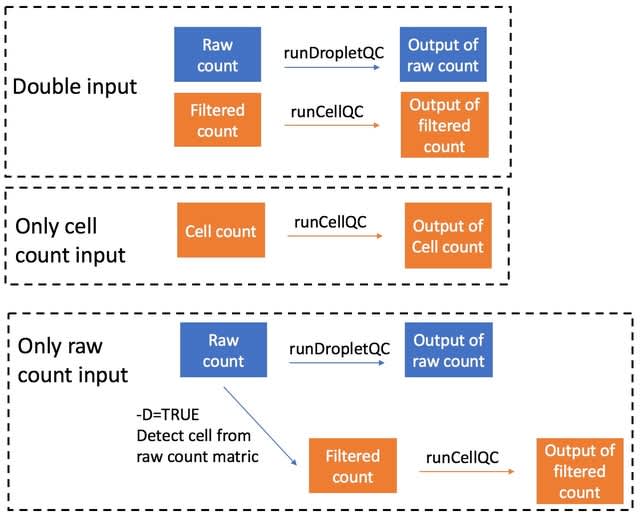
□ Interactive analysis of single-cell data using flexible workflows with SCTK2.0
>> https://www.biorxiv.org/content/10.1101/2022.07.13.499900v1.full.pdf
SCTK enables importing data from the following tools: CellRanger, Optimus, DropEst, BUStools, Seqc, STARSolo and Alevin. In all cases, SCTK parses the standard output directory structure from the pre-processing tools and automatically identifies the count files to import.
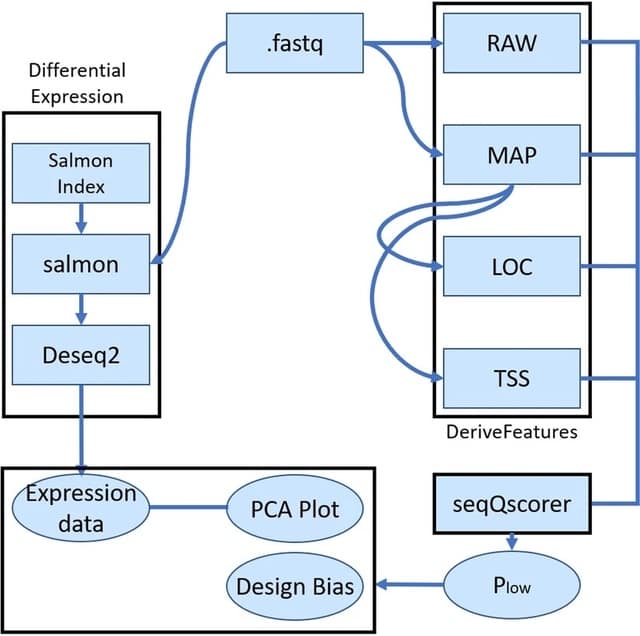
□ seqQscorer: Batch effect detection and correction in RNA-seq data using machine-learning-based automated assessment of quality
>> https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04775-y
seqQscorer detects batch effects in the data. Taken as a confounding factor to correct the data for the clustering of the samples, the quality evaluation led to results comparable to the reference method that uses the real batch information.
The pearsongamma is the correlation b/n distances and a 0–1-vector. Computing a design bias representing the agreement of Plow to biological groups, utilizing Pearson gamma or “normalized gamma”, to have a positive value b/n zero/one they added one and divided the result by two.
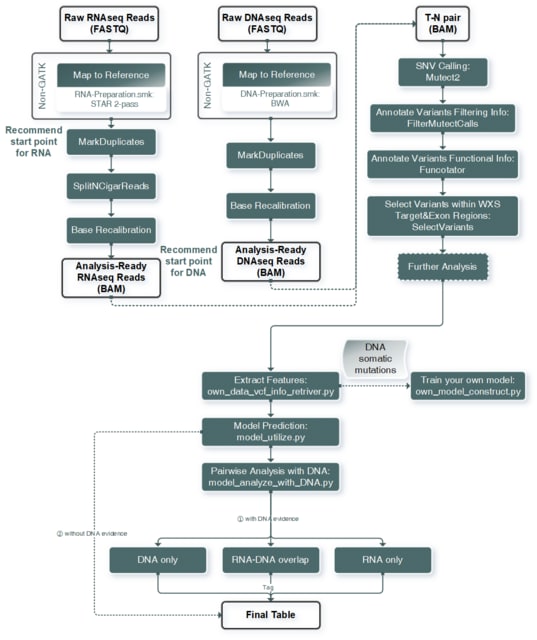
□ RNA-SSNV: A Reliable Somatic Single Nucleotide Variant Identification Framework for Bulk RNA-Seq Data
>> https://www.frontiersin.org/articles/10.3389/fgene.2022.865313/full
The RNA-SSNV is a scalable and efficient analysis method for RNA somatic mutation detection from RNA-WES paired sequencing data which utilized Mutect2 as core-caller and Multi-filtering strategy & Machine-learning based model to maximize precision & recall performance.
RNA-SSNV has a higher functional impact and therapeutic power in known driver genes. Furthermore, VAF (variant allele fraction) analysis revealed that subclonal harboring expressed mutations had evolutional selection advantage and RNA had higher detection power to rescue DNA-omitted mutations.
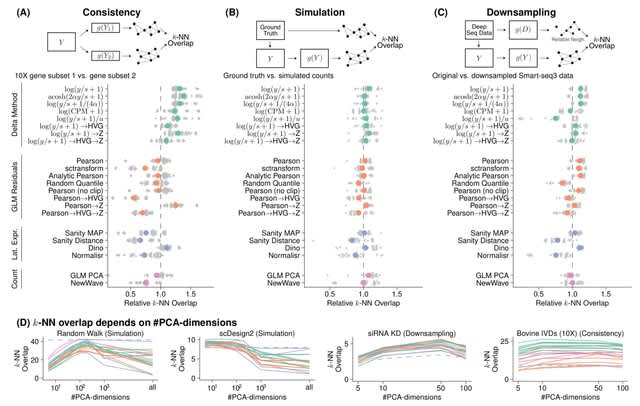
□ Comparison of Transformations for Single-Cell RNA-Seq Data
>> https://www.biorxiv.org/content/10.1101/2021.06.24.449781v3.full.pdf
The Pearson residuals-based transformation has attractive theoretical properties and, in the benchmarks, performed similarly well as the shifted logarithm transformation. It stabilizes the variance across all genes and is less sensitive to variations of the size factor.
Sanity Distance calculates the mean deviation of the posterior distribution of the logarithmic GE; it calculates all cell-by-cell distances, from which it can find the k-NN. Sanity ignores the inferred uncertainty and returns the maximum of the posterior as the transformed value.

□ Recommendations for clinical interpretation of variants found in non-coding regions of the genome
>> https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-022-01073-3
Recommendations aim to increase the number and range of non-coding region variants that can be clinically interpreted, which, together with a compatible phenotype, can lead to new diagnoses and catalyse the discovery of novel disease mechanisms.
Rethinking the standard ‘coding first’ strategy for genetic testing of many genes and conditions, not only through WGS, but also by expanding the regions captured by targeted panels to incl. standardised community-defined regulatory elements, where these remain more appropriate.
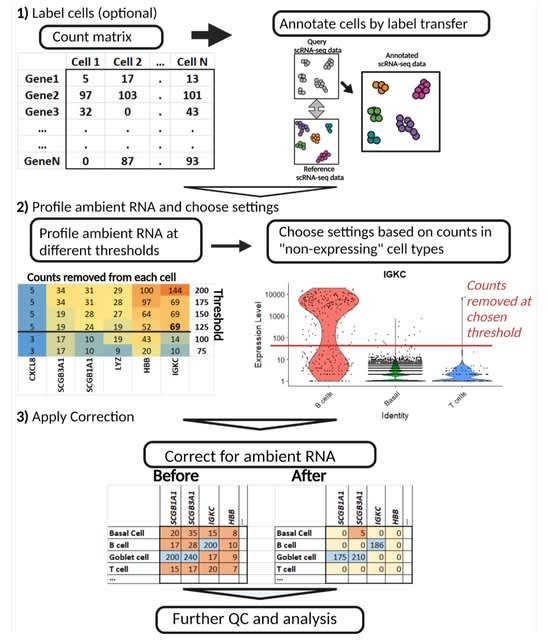
□ FastCAR: Fast Correction for Ambient RNA to facilitate differential gene expression analysis in single-cell RNA-sequencing datasets
>> https://www.biorxiv.org/content/10.1101/2022.07.19.500594v1.full.pdf
Fast Correction for Ambient RNA (FastCAR), a computationally lean and intuitive correction method, optimized for sc-DGE analysis of scRNA-Seq datasets generated by droplet-based methods including the 10XGenomics Chromium platform.










※コメント投稿者のブログIDはブログ作成者のみに通知されます